




Las miopatías inflamatorias idiopáticas (MII) constituyen un grupo heterogéneo de enfermedades que comprometen la musculatura esquelética y se manifiestan por debilidad y signos inflamatorios en la biopsia muscular. El objetivo de este estudio es hacer una caracterización epidemiológica de una cohorte de pacientes con MII en una población del suroccidente colombiano.
MetodologíaDe forma retrospectiva, se revisaron las historias clínicas de pacientes con diagnóstico de MII que fueron tratados en un hospital de cuarto nivel de complejidad en Cali, Colombia, entre el 2011 y el 2017. Se recolectaron variables demográficas, clínicas, serológicas y de tratamiento.
ResultadosSe identificaron 72 pacientes con MII, mayoritariamente mujeres (n=54, 75%). La media de edad al inicio de los síntomas fue de 37,11±19,18 años. Las principales MII fueron dermatomiositis (DM) y polimiositis, las cuales se presentaron en 35 (48,6%) y 25 pacientes (34,7%), respectivamente. Veintiocho pacientes (38,8%) presentaban enfermedad autoinmune asociada, siendo el lupus eritematoso sistémico la más frecuente, al presentarse en 7 (9,72%) pacientes. La biopsia de músculo se realizó en 25 pacientes (34,7%), mientras que 28 (38,8%) tenían anticuerpos antinucleares positivos. La mediana de la creatinfosfoquinasa fue de 877,5 mg/dL (163,5-4.358,3). Sesenta y siete pacientes (93,1%) fueron tratados con glucocorticoides y 18 (25%) con rituximab (RTX) como monoterapia o combinado con otro fármaco inmunosupresor.
ConclusionesLa DM es la condición clínica más frecuente, es común en mujeres y se presenta en la cuarta década de vida. Los tratamientos con los que más se obtuvo mejoría clínica fueron los glucocorticoides, seguidos del RTX en monoterapia o combinado con otros inmunosupresores.
Idiopathic inflammatory myopathies (IIM) are a heterogeneous group of diseases characterised by skeletal muscle involvement, manifested by weakness and inflammatory signs in the muscle biopsy. The objective of this article is to describe the clinical, laboratory, and treatment features of a cohort of patients with IIM in southwest Colombia.
MethodsA retrospective review was conducted on the medical records of patients diagnosed with IIM treated at a fourth-level complexity hospital in Cali, Colombia, from 2011 to 2017. Demographic, clinical, serological, and treatment data were collected.
ResultsA total of 72 patients with IIM were identified, mostly women (n=54, 75%). The mean age at onset of symptoms was 37.11±19.18 years. The main subtypes of IIM were dermatomyositis (DM) and polymyositis, occurring in 35 patients (48.6%) and 25 patients (34.7%), respectively. Twenty-eight patients (38.8%) had associated autoimmune disease, with systemic lupus erythematosus being the most frequent in 7 (9.72%) patients. Muscle biopsy was performed in 25 patients (34.7%), while 28 (38.8%) had positive antinuclear antibodies. The median creatine phosphokinase was 877.5mg/dL (163.5-4358.3). Sixty-seven patients (93.1%) were treated with glucocorticoids, and 18 (25%) patients were treated with rituximab (RTX) as monotherapy or combined with another immunosuppressant drug.
ConclusionsDM is the most frequent subtype of IIM, being common in women and occurring in the fourth decade of life. The most used treatments were glucocorticoids, followed by RTX monotherapy, or combined with other immunosuppressants.
Las miopatías inflamatorias idiopáticas (MII) son un grupo heterogéneo de entidades poco frecuentes, caracterizadas por un compromiso multisistémico que produce inflamación no supurativa de la musculatura esquelética que se puede acompañar de afectación orgánica en piel, articulaciones, pulmón, tracto gastrointestinal y corazón1. Estas condiciones pueden conllevar un deterioro severo de la calidad de vida2. Su incidencia y prevalencia varía entre 1,16 y 19 millones por año y de 2,4 a 33,8 casos por cada 100.000 habitantes, respectivamente, dependiendo de la zona geográfica, los métodos de investigación utilizados y los criterios de clasificación aplicados3.
Las entidades que hacen parte del grupo de MII incluyen la dermatomiositis (DM), la polimiositis (PM), la miopatía necrosante inmunomediada (MNI) y la miositis por cuerpos de inclusión (MSI)1. El síntoma cardinal es la debilidad muscular, aunque puede haber una afectación órgano-específica que produzca un síndrome clínico específico. La demostración serológica de afectación inflamatoria muscular la constituye la elevación de la creatinfosfoquinasa (CPK), la aldolasa, la deshidrogenasa láctica (LDH) y las transaminasas. Entre las ayudas diagnósticas para demostrar el compromiso miopático se encuentran la electromiografía y la imagen por resonancia magnética, la cual, de igual forma, ayuda a orientar el sitio de la biopsia. Esta última es importante pues los hallazgos histopatológicos específicos permiten diferenciar los subtipos clínicos de MII4.
Los criterios de clasificación más utilizados para MII son los de Bohan y Peter5 y los de Tanimoto et al.6, los cuales se basan en hallazgos clínicos, histopatológicos y neurofisiológicos, en combinación con niveles séricos elevados de enzimas musculares, así como autoanticuerpos específicos (solo en Tanimoto et al.). Sin embargo, no incorporan otros hallazgos útiles para distinguir los subtipos de pacientes con miositis, como lo son las imágenes por resonancia magnética y la caracterización por inmunohistoquímica, que incluye la expresión de moléculas del complejo mayor de histocompatibilidad (CMH) en las fibras musculares y subtipos de células inflamatorias involucradas4.
Hasta el momento en Colombia existen pocas descripciones de pacientes con MII, sin embargo, esta es la serie más grande hasta el momento. En este estudio se describen las características clínicas, serológicas y de tratamiento de pacientes con diagnóstico de MII en un hospital de cuarto nivel en Cali, Colombia.
MétodosPacientesDe forma retrospectiva, se revisaron las historias clínicas de pacientes con diagnóstico de MII, según los códigos CIE-10, que fueron tratados en la Fundación Valle del Lili, un hospital de cuarto nivel de complejidad, ubicado en Cali, Colombia, entre el 1 de enero del 2011 y el 31 de diciembre del 2017. Se seleccionaron aquellos pacientes con MII que cumplían criterios clasificatorios de Bohan y Peter5, en tanto que se excluyeron los pacientes menores de 18 años y aquellos con diagnóstico de miopatías por causa tóxica, metabólica, infecciosa o neuromuscular.
Se registraron las características clínicas y de laboratorio al momento del diagnóstico y durante el seguimiento. Las enfermedades autoinmunes asociadas fueron definidas de acuerdo con los criterios clasificatorios para las respectivas enfermedades (artritis reumatoide, lupus eritematoso sistémico, esclerosis sistémica, síndrome de Sjögren, vasculitis). La refractariedad al tratamiento se definió como una falla en la respuesta con 2 medicamentos (de manera individual o recibidos simultáneamente) inmunomoduladores o inmunosupresores al ser administrados en su dosis máxima por 3 meses. La remisión de la enfermedad se definió como la normalización de los valores de CPK y la mejoría de la debilidad muscular por un periodo continuo de 6 meses o más. Este estudio fue aprobado por el comité de ética de nuestra institución.
Análisis estadísticoSe llevó a cabo un análisis estadístico descriptivo. Las variables continuas se expresaron en promedio y desviación estándar o mediana y rango intercuartílico, y fueron analizadas por medio de t test o Mann-Whitney U test, de acuerdo con la distribución de la normalidad. Para el análisis de datos se empleó el programa STATA 12.1.
ResultadosDurante el periodo de 2011-2017 se atendieron 270 pacientes con diagnóstico de miopatía. Después de la revisión de historias clínicas se descartaron 198 pacientes de acuerdo con los criterios de exclusión. Se obtuvo un total de 72 pacientes con MII, de los cuales 18 (25%) fueron hombres y 54 (75%) mujeres, con predominio del sexo femenino en proporción 3:1. La media de edad de los pacientes en el momento del inicio de los síntomas fue de 37,11±19,18 años. En torno a la distribución de MII, 35 pacientes (48,6%) tenían DM, 25 (34,7%) PM, 8 (11,1%) dermatomiositis juvenil, 2 (2,7%) MSI, 1 (1,3%) MNI y 1 (1,3%) miopatía inflamatoria no especificada.
Con respecto a la frecuencia de los síntomas y los signos, la mayoría de los pacientes incluidos (72%) presentó síntomas de debilidad muscular y 17 (23,6%) presentaron disfagia. Dentro de las manifestaciones dermatológicas, las pápulas de Gottron se presentaron en 21 pacientes (29,7%), el signo de Gottron en 9 (12,5%), el rash en heliotropo en 10 (13,8%) y el signo de Chal en 3 (4,1%). Por otra parte, en relación con las comorbilidades, 25 pacientes (34,7%) presentaban enfermedad autoinmune asociada. El lupus eritematoso sistémico (LES), la más frecuente, se presentó en 7 pacientes (9,7%), seguido de la esclerodermia, en 6 (8,3%); la artritis reumatoide, en 5 (6,94%); el síndrome de Sjögren, en 3 (4,2%); la vasculitis del sistema nervioso central, la vasculitis cutánea leucocitoclástica, el síndrome poliglandular autoinmune y el pioderma gangrenoso, en un paciente (1,4%) cada una. Adicionalmente, 39 pacientes (54,17%) presentaron comorbilidades no autoinmunes. Sin embargo, 19 (26,38%) registraron más de 2 enfermedades no autoinmunes de forma concomitante, entre las cuales destacaron: hipertensión arterial, en 11 casos (15,28%), enfermedad tiroidea, en 10 (13,89%) y diabetes mellitus en 6 (8,33%).
En 3 pacientes se documentaron enfermedades neoplásicas: 2 malignidades hematológicas (síndrome mieloproliferativo y linfoproliferativo) y un carcinoma de tiroides. En 14 pacientes (19%) se presentó compromiso pulmonar intersticial. De estos, 5 presentaron patrón en vidrio esmerilado (35,7%), al igual que panalización, de los cuales uno tenía sobreposición con esclerodermia y otro presentó la combinación de ambos patrones. En los casos restantes se describió un patrón restrictivo pulmonar. De estos casos vale la pena mencionar que presentaban sobreposición con esclerodermia y en un caso con escoliosis. Así mismo, de los pacientes con compromiso pulmonar, la mayoría (n=6, 42,8%) alcanzó la remisión clínica en el último control por reumatología, mientras que uno (7,1%) requirió oxígeno domiciliario permanente, otro (7,1%) requirió terapia de rehabilitación pulmonar y el resto presentó algún grado de compromiso clínico persistente, como disnea.
A 25 pacientes (34,7%) se les realizó la biopsia de músculo, lo que ayudó a confirmar el diagnóstico. En los 47 pacientes restantes (65,3%) el diagnóstico fue apoyado en electromiografía con patrón miotático, resonancia nuclear magnética con evidencia de edema muscular y el criterio clínico del reumatólogo. En cuanto a los exámenes de laboratorio, 28 pacientes (38,8%) tenían anticuerpos antinucleares (ANA) positivos (definidos como un título ≥1:160), siendo el patrón moteado el más frecuente, al presentarse en 14 pacientes (19,4%). El anti-Jo1 fue positivo en solo un paciente. Por último, la velocidad de sedimentación globular (VSG) y la proteína C reactiva (PCR) estaban elevadas en 33 (45,8%) y 26 (36%) pacientes, respectivamente. En la tabla 1 se resumen las características clínicas y serológicas de los pacientes con MII.
Características clínicas, demográficas y serológicas
| Características | n (%)=72 |
|---|---|
| Sexo femenino, n (%) | 54 (75%) |
| Edad al inicio de los síntomas, años* | 37,11±19,18 |
| Tipo de miopatía, n (%) | |
| Dermatomiositis | 35 (48,61) |
| Polimiositis | 25 (35%) |
| Dermatomiositis juvenil | 8 (11,11) |
| Miopatía por cuerpos de inclusión | 2 (2,78 |
| Miopatía necrosante | 1 (1,39) |
| Miopatía inflamatoria no especificada | 1 (1,39) |
| Enfermedad autoinmune asociada, n (%) | |
| Lupus eritematoso sistémico | 7 (9,72) |
| Esclerodermia | 6 (8,33) |
| Artritis reumatoidea | 5 (6,94) |
| Síndrome de Sjögren | 3 (4,17) |
| Otras | 4 (5,55) |
| Características clínicas, n (%) | |
| Debilidad muscular simétrica | 52 (72%) |
| Disfagia | 17 (23,61) |
| Rash en heliotropo | 10 (13,89) |
| Pápulas de Gottron | 21 (29,17) |
| Signo de Gottron | 9 (12,5) |
| Signo de Chal | 3 (4,17) |
| Características serológicas, n (%) | |
| Positividad de ANA | 28 (38,8%) |
| Moteado | 14 (19,44) |
| Homogéneo | 8 (11,11) |
| Granular | 3 (4,17) |
| Nucleolar | 3 (4,17) |
| CPK al momento del diagnóstico (mg/dL)** | 877,5 (163,5-4.358,3) |
| Aldolasa al diagnóstico** | 6,7 (0-27,3) |
| AST al diagnóstico** | 57,35 (29,8-177,7) |
| ALT al diagnóstico** | 47,5 (26,2-125,3) |
| LDH al diagnóstico** | 376 (270-618,5) |
| Diagnóstico por biopsia | 25 (34,72) |
| Criterios Bohan y Peter*** | |
| Definitivo | 22 (30,5) |
| Probable | 25 (34,7) |
| Posible | 18 (25) |
| Remisión de la enfermedad | 34 (47,22) |
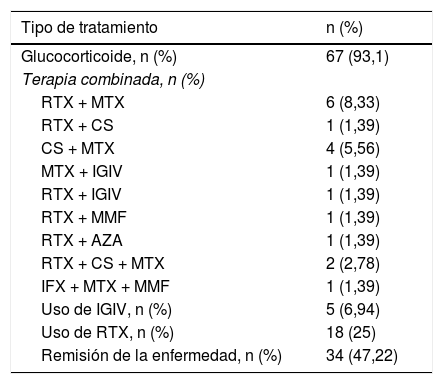
En relación con el tratamiento recibido, la mayoría de los pacientes (n=67, 93,1%) se trató con glucocorticoides. La terapia inmunomoduladora constituye una alternativa para ahorrar dosis de estos. En general, los pacientes recibieron diferentes terapias convencionales inmunomoduladoras en el curso de la enfermedad, como metotrexato en 34 pacientes (47,2%) y azatioprina en 23 (31,9%). No obstante, la respuesta clínica se evidenció en 18 pacientes (25%) tratados con rituximab (RTX) como monoterapia o combinado con otro fármaco inmunosupresor (metotrexato, 6 (8,3%); ciclosporina, uno (1,3%); inmunoglobulina intravenosa, uno (1,3%); azatioprina, uno (1,3%); y micofenolato de mofetilo, uno (1,3%). Trece de los pacientes tratados con RTX presentaron respuesta al tratamiento, con una mediana de tiempo de 60 días (RIC: 36-141) posteriores a la aplicación de la primera dosis del medicamento, con objetivación de una mejoría en la fuerza muscular y descenso en las enzimas musculares. Previamente a la aplicación del RTX, la mediana de CPK fue de 687 U/L (RIC: 205-3.368), y después de su aplicación, teniendo en cuenta el reporte más cercano a la fecha de la evaluación de la fuerza muscular, la mediana de CPK fue de 167 U/L (RIC: 81,5-506,5).
La terapia combinada más utilizada después del RTX-metotrexato fue la ciclosporina-metotrexato, que se empleó en 4 pacientes (5,56%), en tanto que se utilizó inmunoglobulina intravenosa en 5 de ellos (5,64%). Si bien el RTX fue ampliamente utilizado, como monoterapia o en conjunto con otro medicamento, no se empleó como primera línea de tratamiento en ninguno de los casos, a diferencia de los glucocorticoides que se usaron inicialmente en todos los pacientes. En 34 pacientes (47,2%) se obtuvo la remisión de la enfermedad (tabla 2).
Tratamiento
| Tipo de tratamiento | n (%) |
|---|---|
| Glucocorticoide, n (%) | 67 (93,1) |
| Terapia combinada, n (%) | |
| RTX + MTX | 6 (8,33) |
| RTX + CS | 1 (1,39) |
| CS + MTX | 4 (5,56) |
| MTX + IGIV | 1 (1,39) |
| RTX + IGIV | 1 (1,39) |
| RTX + MMF | 1 (1,39) |
| RTX + AZA | 1 (1,39) |
| RTX + CS + MTX | 2 (2,78) |
| IFX + MTX + MMF | 1 (1,39) |
| Uso de IGIV, n (%) | 5 (6,94) |
| Uso de RTX, n (%) | 18 (25) |
| Remisión de la enfermedad, n (%) | 34 (47,22) |
AZA: azatioprina; CS: ciclosporina; IFX: infliximab; IGIV: immunoglobulina intravenosa; MMF: micofenolato de mofetilo; MTX: metrotexate; RTX: rituximab.
Este es el registro más grande de pacientes con MII en Colombia. El estudio se realizó con el objetivo de analizar de forma retrospectiva las características clínicas, de laboratorio y tratamiento de los pacientes con MII en un hospital en Cali, Colombia.
En torno al subtipo de MII, la entidad más frecuente en casi la mitad de los pacientes fue la DM, a diferencia de otros estudios en los cuales la PM fue la entidad más común7,8. Este hallazgo podría estar relacionado con que varios de los pacientes durante el proceso diagnóstico fueron reclasificados como DM después de los hallazgos histopatológicos compatibles con esta entidad. La biopsia muscular se realizó en el 35% de la población, constituyendo una herramienta adicional para el diagnóstico certero de los diferentes subgrupos de MII9-11.
Las MII se asocian a diversas enfermedades autoinmunes y se describen con una frecuencia muy variable, entre el 7% y el 60% de acuerdo con diferentes series12,13. En nuestra población la asociación con enfermedades autoinmunes fue del 35%, principalmente con LES. En un estudio retrospectivo realizado en Brasil con una cohorte de 220 pacientes, en el cual la intención fue estudiar la sobreposición de síndromes (PM/DM), se observó que las asociaciones más comunes fueron la esclerosis sistémica, el LES y la AR, con frecuencias del 48,4%, 29% y 22,6%, respectivamente14. En una cohorte de 160 pacientes con MII en Hungría, de aquellos pacientes que tenían un síndrome de sobreposición (39), 33,3% tenía escleromiositis, 27,6% AR, 23,1% síndrome de Sjögren y 12,8% LES15.
Los ANA pueden ser positivos en un 5,9% y hasta un 30,8% de la población sana16. Se presentan con mayor frecuencia en mujeres y en individuos de edad avanzada17. Pueden ser positivos también en enfermedades autoinmunes como AR, síndrome de Sjögren, esclerosis sistémica, enfermedad mixta del tejido conectivo y MII. En nuestra población, el 38,8% de los pacientes tenía ANA positivos, siendo el patrón moteado el más frecuente. Por otra parte, el anti-Jo1 fue positivo en solo un paciente. Este anticuerpo hace parte del grupo de anticuerpos específicos de miositis (MSA) y es el que se presenta con mayor frecuencia (hasta 20%) en MII. Además, se asocia con el síndrome antisintetasa y constituye un marcador de mal pronóstico18. El resto del panel de autoanticuerpos específicos no se evaluó puesto que estos no se encuentran disponibles en nuestra institución.
La asociación de MII y malignidad se reportó desde 191619. Una de las cohortes más grandes de MII de Australia incluyó a 537 pacientes con diagnóstico confirmado por biopsia, y se encontraron 116 casos de malignidad en 104 pacientes, con una asociación mayor en aquellos pacientes con DM20. Un metaanálisis reciente describe el aumento del riesgo para malignidad en pacientes con PM y DM, con un riesgo relativo (RR) de 1,62 (IC 95% 1,19-2,04) y 5,50 (IC 95% 4,31-6,70), respectivamente21. Sin embargo, en nuestro estudio, solo 3 pacientes presentaron enfermedades neoplásicas asociadas con miopatía inflamatoria.
En relación con el tratamiento, son 3 los pilares fundamentales, e incluyen el control o la eliminación de factores causales, el uso de terapia inmunosupresora y rehabilitación. En torno a los inmunosupresores, los GC constituyen la primera línea de tratamiento. En nuestro estudio, casi la totalidad de los pacientes recibió esta medicación. El metotrexate, la ciclosporina, la azatioprina, el micofenolato mofetilo, la ciclofosfamida y el IVIG constituyen una alternativa para aquellos casos refractarios, o son utilizados como ahorradores de dosis de GC. Por otra parte, en pacientes refractarios a la primera línea de tratamiento, tras descartar otra condición subyacente (como miopatía necrosante o miositis por cuerpos de inclusión), se considera el inicio de agentes biológicos como el RTX22, el cual puede ser utilizado en conjunto con otro fármaco inmunomodulador o en monoterapia con GC.
El RTX es un anticuerpo monoclonal que se une al antígeno CD20 expresado en la superficie de los linfocitos B, lo que da como resultado la depleción de los CD20 positivos en sangre periférica dentro de los siguientes 6-9 meses23. Su uso se implementó de manera empírica en pacientes que no presentaron una adecuada respuesta al tratamiento convencional, bajo la premisa y evidencia de autoanticuerpos circulantes en alrededor de un 80% en los pacientes con MII y de células B en la región perivascular de los músculos en este grupo de pacientes24-26. En nuestro estudio 18 pacientes recibieron este tipo de terapia inmunomoduladora combinada con RTX, siendo el metotrexato el fármaco más utilizado junto al RTX. Se observó una tasa de remisión de aproximadamente el 40%, en comparación con otros estudios cuya tasa es reportada como menor al 10%8.
Dentro de las limitaciones del estudio, no fue posible hacer la biopsia muscular en la totalidad de los pacientes, teniendo en cuenta que no se llevaba a cabo de manera protocolaria desde la fecha en que se empezó a registrar a los pacientes. Otra limitación incluye el posible sesgo de selección al ser un estudio retrospectivo, e incluir pacientes registrados de acuerdo con el código CIE-10.
ConclusiónEn esta cohorte, la miopatía inflamatoria más prevalente fue la DM, que es común en mujeres y se presenta en la cuarta década de vida. El tratamiento con glucocorticoides fue el más utilizado. La terapia inmunosupresora combinada constituye una alternativa para el control de la enfermedad.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
