








La enfermedad de Pompe o glucogenosis tipo II es una enfermedad poco frecuente causada por mutaciones en el gen GAA que conduce a la deficiencia de la enzima alfa-1,4-glucosidasa ácida. Como resultado del defecto enzimático se produce una acumulación progresiva de glucógeno intralisosomal en diversos tejidos, causando afectación del músculo liso, cardíaco y esquelético. Cuando la edad de inicio de la enfermedad es posterior al primer año de vida se denomina enfermedad de Pompe de inicio tardío (LOPD) y tiene como principales manifestaciones clínicas la debilidad de la musculatura axial y proximal de cinturas y la disfunción respiratoria. El tratamiento estándar con la terapia enzimática sustitutiva (ERT), disponible hace más de 15 años, consigue cambiar el curso de la enfermedad, aunque la eficacia del tratamiento se ve reducida con el paso del tiempo. Las nuevas terapias enzimáticas suponen nuevas oportunidades de tratamiento para los pacientes con LOPD. Este trabajo presenta recomendaciones actualizadas de un grupo de expertos en la enfermedad de Pompe sobre el diagnóstico, tratamiento y seguimiento de los pacientes con LOPD, con el objetivo de proporcionar una guía para el manejo clínico de la enfermedad.
Pompe disease or glycogenosis type II is a rare disease caused by mutations in the GAA gene that leads to deficiency of the acid alpha-1,4-glucosidase enzyme. As a result of the enzymatic defect, a progressive accumulation of intralysosomal glycogen occurs in various tissues, causing smooth, cardiac and skeletal muscle involvement. When the age of onset of the disease is after the first year of life, it is called late-onset Pompe disease (LOPD). Weakness of the axial and proximal waist muscles and respiratory dysfunction are common manifestations. Enzyme replacement therapy (ERT) has been available for more than 15 years and is the standard treatment. This therapy changes the course of the disease, although the effectiveness of the treatment reduces over time. New enzyme therapies represent new treatment opportunities for patients with LOPD. Here we present updated recommendations from a group of experts in Pompe disease on the diagnosis, treatment and follow-up of LOPD patients, with the aim of providing a guide for the clinical management of the disease.
La enfermedad de Pompe o glucogenosis tipo II (GSDII, por sus siglas en inglés Glycogen Storage Disease type II), es una enfermedad de origen genético de herencia autosómica recesiva. Se trata de una entidad poco frecuente, producida por el déficit de la enzima alfa-1,4-glucosidasa ácida (GAA), codificada por el gen GAA, situado en el brazo largo del cromosoma 17 (17q25.3). La enzima GAA es la encargada de hidrolizar el glucógeno intralisosomal, y su ausencia o deficiencia conduce a la acumulación progresiva de glucógeno en diversos tejidos, si bien los síntomas se deben fundamentalmente a la afectación del músculo esquelético, liso y/o cardiaco1,2.
La enfermedad de Pompe se clasifica en 2 grupos en función de la edad de aparición de los síntomas y la presencia de cardiomiopatía. La enfermedad de Pompe infantil clásica se produce cuando la actividad enzimática de GAA está ausente o es muy reducida, en general menor al 1% (IOPD, por sus siglas en inglés Infantil Onset Pompe Disease). La enfermedad de Pompe de inicio tardío (LOPD, por sus siglas en inglés Late Onset Pompe Disease) se produce cuando la actividad de GAA está reducida, siendo mayor al 1 y menor del 30%3.
Los pacientes con LOPD desarrollan los síntomas durante el primer año de vida y, si no son tratados, raramente sobreviven más de 18 meses. Las principales manifestaciones clínicas incluyen hipotonía, debilidad muscular generalizada, disfagia, insuficiencia respiratoria progresiva, miocardiopatía hipertrófica, hepatomegalia y macroglosia1,4. Se han descrito pacientes con un inicio de síntomas de la enfermedad durante el primer año de vida, pero sin miocardiopatía hipertrófica asociada, y estos casos se han denominado formas infantiles no clásicas1,4. Aquellos pacientes que manifiestan los síntomas a partir del primer año de vida, y que no se acompañan de afectación cardiaca relevante, se clasifican como LOPD. Estos pacientes pueden iniciar síntomas en la edad infantil, juvenil o a lo largo de la vida adulta. Las principales manifestaciones clínicas de los pacientes con LOPD incluyen debilidad muscular de distribución axial y proximal de cinturas de curso progresivo con afectación temprana de la musculatura respiratoria. Los primeros síntomas, antes de la aparición de la debilidad, pueden ser inespecíficos e incluyen intolerancia al ejercicio y fatiga. En esta fase, la mayoría de los pacientes presentan cifras elevadas de creatina-cinasa (CK) de manera persistente, lo que puede facilitar un diagnóstico temprano de la enfermedad. El deterioro motor progresa de forma lenta y se acompaña de insuficiencia respiratoria, lo que conduce hacia la pérdida de la marcha autónoma y la necesidad de ventilación mecánica. Los problemas respiratorios son la principal causa de morbimortalidad de la enfermedad y en muchas ocasiones preceden a la pérdida de la deambulación autónoma4.
El objetivo de este trabajo es revisar y actualizar las recomendaciones existentes sobre el diagnóstico, tratamiento y seguimiento de los pacientes con enfermedad de Pompe de inicio tardío, con el fin de incorporar los nuevos conocimientos derivados de la experiencia acumulada tras más de 15 años de terapia enzimática sustitutiva (ERT, por sus siglas en inglés enzyme replacement therapy) y la aparición en los últimos años de nuevas terapias enzimáticas5–11. La publicación de recomendaciones actualizadas tiene el objetivo de facilitar y homogeneizar en lo posible el manejo clínico de la enfermedad entre los profesionales implicados en su cuidado.
Material y métodosLas presentes recomendaciones son el resultado de la revisión de las guías y recomendaciones existentes en la bibliografía5–11 y de un panel Delphi para conseguir el mayor consenso de un equipo multidisciplinar de 19 expertos compuesto por especialistas en neumología, neuropediatría, enfermedades metabólicas hereditarias, neurología y medicina interna. Se realizaron 3 cuestionarios, con un total de 38 preguntas sobre el diagnóstico, tratamiento y seguimiento de la LOPD, que fueron contestados electrónicamente por los expertos. Aquellos puntos donde no se alcanzó un acuerdo con el cuestionario inicial, fueron discutidos en una reunión telemática hasta llegar a un consenso en las recomendaciones finales.
Resultados y discusiónDiagnósticoDado que la enfermedad de Pompe es una enfermedad tratable que puede conducir a una muerte prematura, es de gran importancia alcanzar un diagnóstico temprano. Este diagnóstico puede ser complejo dada la heterogeneidad clínica que caracteriza las formas de inicio tardío9.
Análisis de la actividad de la enzima alfa-1,4-glucosidasa ácidaAnte la sospecha clínica de enfermedad de Pompe, la prueba de cribado recomendada es el análisis de la actividad de la enzima GAA en sangre seca impregnada en papel (DBS, por sus siglas en inglés dry blood spot)9. La sospecha de enfermedad de Pompe se basa en la conjunción de los síntomas y signos clínicos compatibles con la enfermedad, que incluyen niveles elevados de la actividad CK en suero, presencia de debilidad muscular progresiva, en especial debilidad de cinturas o axial, y/o afectación respiratoria de origen neuromuscular6.
La determinación de la actividad enzimática en DBS es una prueba rápida, accesible y altamente sensible, sin embargo, puede dar falsos positivos, por lo que es necesario confirmar el resultado cuando se obtiene una reducción de la actividad enzimática6. Cabe destacar que los portadores de una única variante (heterocigotos de una mutación del gen GAA) también pueden presentar actividad enzimática en el rango patológico, tal como han demostrado algunos estudios12,13, así que es recomendable interpretar los resultados con precaución.
El estudio electromiográfico puede mostrar un patrón miopático pudiendo identificarse descargas miotónicas, especialmente en musculatura paraespinal, aún en ausencia de miotonía clínica. Sin embargo, la presencia de miotonía eléctrica no es específica de la enfermedad de Pompe y puede verse en otras enfermedades musculares como en la distrofia miotónica tipo I y II, canalopatías, miopatías miofibrilares y tóxicas, entre otras4,6,14.
Para confirmar el diagnóstico debe realizarse un estudio genético10. A la hora de planificar el estudio genético debe tenerse en cuenta que además de cambios puntuales en la secuencia del gen GAA, también se han identificado grandes deleciones, duplicaciones y variantes intrónicas profundas. Por tanto, en caso de que la secuenciación directa (mediante estudio Sanger o NGS) no sea concluyente, son necesarios estudios que permitan detectar ganancias o pérdidas de material genético, también en regiones intrónicas3.
Una buena guía para valorar la patogenicidad de las variantes encontradas en el GAA es la base de datos del Erasmus Center: https://www.pompevariantdatabase.nl; que contiene 911 variantes del gen GAA identificadas, de las cuales 648 (71%) son patogénicas y de estas, en 336 se conoce su correlación con el fenotipo (fecha de consulta 18 de abril 2024)15,16. Cuando un estudio genético completo no permite identificar variantes bialélicas patogénicas y la sospecha de enfermedad de Pompe es muy elevada, el análisis de la actividad GAA en diferentes tejidos puede ser de ayuda para apoyar el diagnóstico8. Se considera que para analizar la actividad GAA, el análisis enzimático en fibroblastos cultivados obtenidos por biopsia de piel es la prueba estándar, sin embargo, es un procedimiento algo invasivo y los resultados pueden tardar hasta 6 semanas. Hay estudios que indican que el análisis enzimático en sangre proporciona resultados comparables a los obtenidos con fibroblastos, y además es un método menos invasivo y más rápido14,17. El estudio enzimático puede también realizarse en músculo esquelético si se dispone de tejido para realizar el mismo.
Estudio genético a los familiares en riesgoSe recomienda realizar asesoramiento genético a los familiares del paciente y realizar estudios genéticos a los familiares en riesgo de desarrollar la enfermedad o de ser portadores. Cuando los familiares en riesgo son menores de edad es recomendable discutir con los progenitores o representantes legales la necesidad del estudio genético. La detección de portadores de las variantes patogénicas facilita la planificación familiar10.
Resonancia magnética muscularPara evaluar la distribución y grado de afectación muscular en la LOPD se puede utilizar la imagen por resonancia magnética (RM) muscular, utilizando secuencias de tipo T1, que permiten identificar reemplazo muscular por tejido graso, y secuencias STIR, que permiten visualizar edema en la musculatura afecta. Las secuencias de T1 muestran un patrón de reemplazo graso característico útil para el diagnóstico y pueden ayudar a seleccionar qué músculo biopsiar, en el caso de que una biopsia sea necesaria4. El patrón característico en T1 se caracteriza por reemplazo graso de los siguientes músculos: lengua, subescapular, musculatura paraespinal y abdominal, glúteo medio y menor, musculatura posterior del muslo, aductor mayor y vasto intermedio18,19. Característicamente, la musculatura distal de las piernas se encuentra respetada. Se ha demostrado que existe una buena correlación entre la fuerza muscular, las escalas motoras funcionales y el grado de reemplazo graso en el tejido muscular en la RM y que la aparición de los primeros signos de sustitución grasa puede preceder a la debilidad muscular en los pacientes estudiados por elevación de las cifras de CK18. Asimismo, la detección de un incremento progresivo en el reemplazo de la musculatura esquelética por tejido adiposo puede ser un factor a tener en cuenta para iniciar el tratamiento con ERT en los pacientes paucisintomáticos18.
Biopsia muscularLa biopsia de músculo puede servir para apoyar el diagnóstico de la enfermedad de Pompe. Sin embargo, en las formas LOPD su resultado puede ser inespecífico o incluso normal, especialmente si se toma la biopsia de un músculo no afectado. Se estima que el 30% de los pacientes con LOPD presentan biopsia normal o inespecífica6,9. Es importante la correcta conservación de las muestras y realizar una óptima congelación, fijación, inclusión y tinción20. La tinción con ácido periódico de Schiff (PAS) en sección semifina es el método más fiable para visualizar vacuolas con acúmulo de glucógeno con independencia de su tamaño. Sin embargo, esta técnica no está disponible en todos los laboratorios, y no es específica. Asimismo, la tinción de fosfatasa ácida puede ayudar a identificar el acúmulo de material lisosomal en el sarcoplasma de las fibras musculares, incluso en biopsias sin evidencia clara de presencia de vacuolas ni otros cambios estructurales9,20,21.
Evaluar las potenciales complicaciones vascularesAunque son poco frecuentes, se han documentado alteraciones cardiovasculares y malformaciones vasculares22. Por este motivo después del diagnóstico es recomendable discutir con los pacientes los riesgos potenciales de las complicaciones vasculares y la posibilidad de realizar pruebas diagnósticas para su detección20,23,24.
La angiografía por tomografía computarizada (angioTAC) y la angiografía por resonancia magnética (angioRM) permiten detectar la presencia de anomalías vasculares. Estas pruebas deben cubrir los vasos cerebrales, troncos supraaórticos, la aorta y sus ramas principales. El angioTAC es el método más recomendable, especialmente en los pacientes con insuficiencia respiratoria, al ser más rápido y cómodo. En cambio, cuando los pacientes pueden soportar pruebas de mayor duración es más recomendable la angioRM, debido a su menor irradiación. En caso de detectar un aneurisma intracraneal se debe valorar el riesgo de rotura y de ser necesario iniciar el tratamiento endovascular20.
La afectación cardiaca en LOPD es muy poco frecuente. Un estudio documentó que el 10% de los pacientes presentaban un intervalo P-R corto, el 7% tenían la función sistólica del ventrículo izquierdo reducida y el 5% presentaban una leve elevación de la masa ventricular izquierda, mientras que otro estudio encontró que el 13% de los pacientes presentaban algún grado de hipertrofia del corazón. Asimismo, se han reportado pacientes con LOPD con alteraciones en el ritmo cardíaco, como arritmias sinusales, taquicardia supraventricular, síndrome Wolff-Parkinson-White o bloqueo atrio-ventricular32. El electrocardiograma y el Holter durante 24h permiten descartar alteraciones del ritmo cardíaco como el síndrome Wolff-Parkinson-White, a menudo asociado con un intervalo P-R corto o bloqueo auriculoventricular de segundo grado20, mientras que la ecocardiografía evalúa la función y estructura ventricular4,14.
Diagnóstico de la afectación respiratoria en la enfermedad de PompeLos pacientes con LOPD pueden presentar debilidad de la musculatura respiratoria, incluso antes de que exista debilidad significativa en extremidades. Los problemas respiratorios son la principal causa de morbimortalidad en la enfermedad9,25.
La debilidad progresiva de la musculatura respiratoria lleva a un patrón de respiración superficial y a una mayor carga de trabajo y riesgo de fatiga muscular que conducen a la hipoventilación alveolar, inicialmente nocturna que puede progresar a una hipoventilación también diurna. Asimismo, la debilidad de la musculatura respiratoria, principalmente intercostal y abdominal, causa tos ineficaz a la par que hay una alteración en la protección de la vía aérea por la alteración de la musculatura orofaríngea que contribuye a la tos ineficaz y causa trastornos de deglución y fonación26.
Algunos de los síntomas relacionados con la debilidad muscular respiratoria incluyen la disnea, tanto de esfuerzo como en reposo, una capacidad física reducida, episodios de broncoaspiración e infecciones respiratorias. Asimismo, algunos síntomas asociados con la hipoventilación nocturna incluyen la interrupción del sueño, la hipersomnolencia diurna y la fatiga9,25.
La progresión de la enfermedad y el compromiso diafragmático son muy variables y por lo tanto es de gran importancia evaluar y monitorizar la función respiratoria de los pacientes desde el diagnóstico25,27.
Evaluación inicialSe recomienda que, tras el diagnóstico, los pacientes con enfermedad de Pompe sean evaluados y seguidos en consultas de neumología especializadas integradas en unidades multidisciplinares de atención al paciente neuromuscular. Además de pruebas funcionales respiratorias, se recomienda realizar estudios específicos dirigidos a descartar trastornos respiratorios durante el sueño, que pueden aparecer pronto en la evolución de la enfermedad.
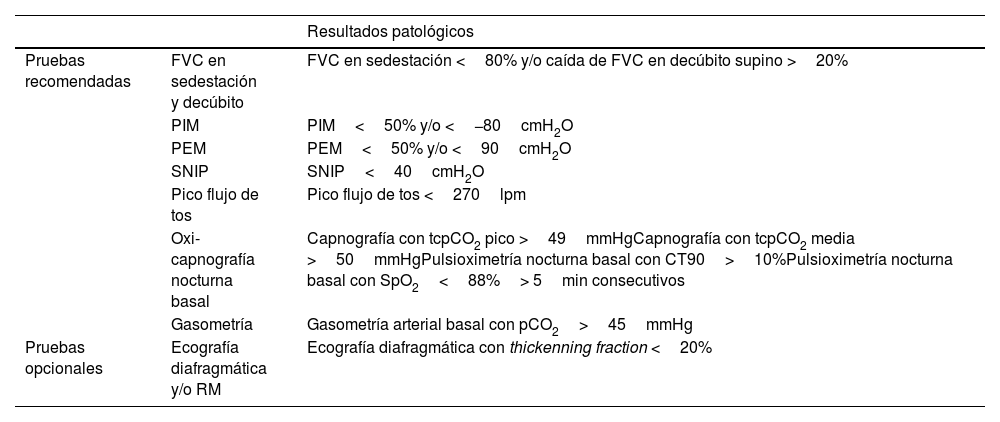
Las pruebas recomendadas incluyen la capacidad vital forzada (FVC), la presión inspiratoria máxima (PIM), la presión espiratoria máxima (PEM), la presión inspiratoria nasal (SNIP), el pico flujo de tos, la oxi-capnografía nocturna basal y la gasometría (tabla 1)25,26.
Pruebas recomendadas para la evaluación inicial del paciente con enfermedad de Pompe y resultados patológicos25,26,28
| Resultados patológicos | ||
|---|---|---|
| Pruebas recomendadas | FVC en sedestación y decúbito | FVC en sedestación <80% y/o caída de FVC en decúbito supino >20% |
| PIM | PIM<50% y/o <−80cmH2O | |
| PEM | PEM<50% y/o <90cmH2O | |
| SNIP | SNIP<40cmH2O | |
| Pico flujo de tos | Pico flujo de tos <270lpm | |
| Oxi-capnografía nocturna basal | Capnografía con tcpCO2 pico >49mmHgCapnografía con tcpCO2 media >50mmHgPulsioximetría nocturna basal con CT90>10%Pulsioximetría nocturna basal con SpO2<88%> 5min consecutivos | |
| Gasometría | Gasometría arterial basal con pCO2>45mmHg | |
| Pruebas opcionales | Ecografía diafragmática y/o RM | Ecografía diafragmática con thickenning fraction <20% |
cmH2O: centímetro de agua; CT90: porcentaje de tiempo con saturación de O2 por debajo del 90%; FVC: capacidad vital forzada; pCO2: presión parcial de dióxido de carbono; PIM: presión inspiratoria máxima; PEM: presión espiratoria máxima; RM: resonancia magnética; SNIP: presión inspiratoria nasal; SpO2: saturación de oxígeno; tcpCO2: presión transcutánea de dióxido de carbono.
La FVC debe evaluarse tanto en sedestación como en decúbito, y permite identificar la presencia de una alteración ventilatoria restrictiva debido a la debilidad muscular inspiratoria. La FVC en sedestación se considera reducida cuando es menor al 80%. Asimismo, un descenso de la FVC mayor al 20% en posición supina respecto la sedestación sugiere la existencia de debilidad diafragmática importante25,26,28.
La PIM y PEM son pruebas que permiten medir la fuerza de los músculos inspiratorios y espiratorios, respectivamente. La PIM mide la máxima presión al realizar una inspiración forzada a partir del volumen residual contra una oclusión (maniobra de Muller). Se considera que la PIM es patológica cuando es menor al 50% del valor esperado y/o mayor a −80cmH2O. La PEM mide la presión máxima al realizar una espiración forzada a partir de la capacidad pulmonar total contra una oclusión (maniobra de Valsalva). La PEM es patológica cuando es menor al 50% del valor esperado y/o menor a 90cm H2O25,26,29.
La SNIP evalúa la presión máxima generada al esnifar con una fosa nasal taponada. Esta prueba es no invasiva y se considera complementaria o una alternativa a la PIM cuando hay debilidad facial o mala oclusión bucal. Una SNIP menor a 40cmH2O se considera patológica25,26.
El pico flujo de tos es una prueba que permite evaluar la eficacia de los músculos espiratorios para eliminar secreciones. Los adultos sanos presentan valores superiores a 350l/min, mientras que un pico flujo de tos menor a 270l/min indica que la tos es ineficaz para eliminar secreciones26.
La oxi-capnografía nocturna basal permite la monitorización no invasiva a tiempo real de la saturación de oxígeno (SpO2) y la presión transcutánea de dióxido de carbono (tcpCO2), lo que informa sobre la ventilación alveolar y permite detectar periodos de hipoventilación nocturna. Se consideran valores patológicos de la capnografía cuando la tcpCO2 pico es mayor a 49mmHg o la tcpCO2 media es mayor a 50mmHg. Asimismo, cuando la SpO2 nocturna está por debajo del 90% más del 10% del tiempo o por debajo del 88% durante más de 5 minutos consecutivos se considera que el paciente está hipoventilado25,26,28.
La gasometría arterial permite determinar el estado de oxigenación, ventilación y ácido-base a partir de las mediciones de la presión parcial de oxígeno (pO2), presión parcial de dióxido de carbono (pCO2), saturación arterial de oxígeno (SaO2) y pH. Se considera hipercapnia diurna cuando la gasometría arterial basal presenta un pCO2 mayor a 45mmHg25,26.
Las pruebas de imagen como la ecografía diafragmática y la RM pueden ayudar a valorar la función diafragmática. La ecografía permite medir el espesor del diafragma y evaluar sus movimientos, mientras que la RM permite evaluar la cinética del diafragma25,26. La ecografía diafragmática y la RM se recomiendan como pruebas complementarias opcionales. El factor de engrosamiento diafragmático (thickenning fraction) se mide como el cambio en el grosor del diafragma entre el final de la inspiración y la espiración respecto al grosor diafragmático al final de la espiración30. Se considera que hay disfunción diafragmática cuando el factor de engrosamiento es menor del 20%31. Por el contrario, en la actualidad no existe un consenso sobre cuáles son los valores normativos de la RM y qué valores deben ser considerados patológicos25.
| Recomendaciones para el diagnóstico de la LOPD |
| • Se recomienda realizar un DBS ante los pacientes con hiperCKemia asintomática, y sospecha de miopatía, especialmente si el paciente presenta debilidad de cintura y/o axial y/o afectación respiratoria de origen neuromuscular. |
| • En los casos en que la actividad enzimática en gota seca esté reducida, se recomienda confirmar este resultado en sangre total y realizar estudio genético. |
| • En los casos de pacientes con actividad enzimática reducida, pero con estudio genético de secuenciación negativo, se recomienda realizar un estudio genético extendido. |
| • La biopsia muscular está recomendada para aquellos casos con sospecha de enfermedad de Pompe y estudio genético negativo. |
| • La resonancia magnética (RM) muscular permite afianzar la sospecha diagnóstica y puede apoyar el inicio del tratamiento. |
| • Ante el diagnóstico establecido de enfermedad de Pompe, se recomienda realizar estudio genético a todos los familiares en riesgo de desarrollar la enfermedad o de ser portadores. En aquellos pacientes menores de edad, se recomienda discutir el caso con sus padres o representantes legales. |
| • Tras el diagnóstico, se recomienda discutir las potenciales complicaciones vasculares de la enfermedad con los pacientes, con el objetivo de realizar pruebas diagnósticas para detectar alteraciones vasculares. |
| Diagnóstico de la afectación respiratoria |
| • El paciente debe ser derivado a neumología desde el momento del diagnóstico, independientemente de sus síntomas. |
| • Las pruebas recomendadas para la evaluación inicial del paciente con enfermedad de Pompe deben incluir FVC en sedestación y decúbito, PIM y PEM, SNIP, pico flujo de tos, oxi-capnografía nocturna basal y gasometría. |
| • La ecografía y/o la resonancia magnética diafragmática son pruebas opcionales complementarias para la evaluación inicial del paciente. |
El tratamiento estándar para la enfermedad de Pompe, disponible desde 2006, es la ERT con alglucosidasa alfa recombinante humana (rhGAA). La ERT se administra en perfusión intravenosa en dosis de 20mg/kg/cada 2 semanas, y está indicada para pacientes con diagnóstico confirmado de enfermedad de Pompe de todas las edades33.
El tratamiento con ERT en los pacientes con LOPD ha demostrado que mejora o estabiliza la función motora y respiratoria, reduce el riesgo de pérdida de deambulación autónoma y prolonga la supervivencia34.
Una vez se ha realizado el diagnóstico de la LOPD, la decisión de iniciar el tratamiento con ERT depende del estado clínico del paciente. En general se recomienda iniciar la ERT de forma inmediata en aquellos pacientes sintomáticos con debilidad muscular en el examen físico o reducción de los parámetros pulmonares en las pruebas de función pulmonar, independientemente del uso de ventilación mecánica no invasiva (VMNI). La presencia de síntomas como las apneas-hipopneas del sueño o la disnea con esfuerzo, se consideran suficientes para iniciar el tratamiento solo si están asociados a datos objetivos de disfunción ventilatoria35.
En cambio, los pacientes asintomáticos, sin signos objetivos de la enfermedad, deben ser monitorizados de manera regular, para iniciar tratamiento en caso de aparición de deterioro clínico de la fuerza muscular o la función pulmonar35. La existencia de una RM muscular que muestre reemplazo progresivo de tejido muscular por tejido adiposo puede considerarse como un factor a tener en cuenta para el inicio del tratamiento18.
Una vez iniciada la ERT se debe monitorizar a los pacientes para evaluar la respuesta al tratamiento35.
Tratamiento domiciliarioLa calidad de vida de los pacientes con LOPD se ve afectada negativamente por la gran inversión de tiempo que deben dedicar a recibir el tratamiento. La terapia enzimática puede ser administrada de forma domiciliaria, lo que aporta una mayor autonomía y flexibilidad al paciente, y tiene un menor impacto en su vida diaria. Un estudio realizado con 121 pacientes con LOPD demostró que la administración domiciliaria de la ERT es segura cuando se establecen unos protocolos adecuados de administración y manejo de las reacciones asociadas con la infusión y se dispone de personal sanitario entrenado36. En consecuencia, se recomienda el tratamiento domiciliario tras confirmar una buena tolerancia a la infusión en el hospital.
Cambio de la terapia de reemplazo enzimáticoAunque más del 90% de los pacientes se benefician del tratamiento con ERT, en general, el beneficio clínico es mayor durante los primeros 3-5 años del mismo, seguido de un lento empeoramiento posterior en al menos la mitad de los casos37. Las razones de la limitada eficacia y poca durabilidad del efecto de la ERT en los pacientes con LOPD aún no están claras, aunque diversos factores pueden estar influyendo; por un lado, el músculo esquelético presenta escasa densidad de receptores manosa-6-fostafo (M6P), lo que limita la captación y entrada del enzima en este tejido, el enzima actúa en medio ácido y, por tanto, tiene su efecto preferente sobre el glucógeno intralisosomal y no sobre el citoplasmático, la disfunción lisosomal y mitocondrial establecida no se corrige con la administración exógena de la enzima deficitaria y puede interferir además con la captación y dirección de la terapia hacia el lisosoma. Estas limitaciones han llevado a la búsqueda de nuevos tratamientos. Una de las estrategias seguidas ha consistido en optimizar la ERT. Actualmente existen 2 nuevas terapias enzimáticas disponibles en el mercado.
Por una parte, avalglucosidasa alfa, es una rhGAA en la que se ha incrementado el número de residuos M6P con elevada afinidad por el receptor de M6P de la superficie celular, mejorando su captación por las fibras musculares38. El estudio COMET, un ensayo clínico fase III multicéntrico multinacional, mostró que avalglucosidasa alfa no era inferior a alglucosidasa alfa presentando mejores resultados clínicos en la función respiratoria y motora. Este estudio incluyó 100 pacientes naïve con LOPD aleatorizados 1:1 para recibir alglucosidasa alfa o avalglucosidasa alfa durante un período de 49 semanas, que se siguió de una fase de extensión abierta en la que todos los pacientes recibieron aval-glucosidasa alfa. En detalle, aquellos pacientes tratados con avalglucosidasa alfa mostraron un mayor incremento a nivel nominal de la FVC (2,89 vs. 0,46%) y recorrieron una mayor distancia en la prueba de la marcha de 6min (6MWT: 32,21 vs. 2,19m) a las 49 semanas de seguimiento comparados con los pacientes que recibieron alglucosidasa alfa. Las diferencias mostradas en el 6MWT resultaron estadísticamente significativas para no inferioridad entre ambas terapias (p=0,0074), pero no se alcanzó la superioridad estadística (p=0,06). Por otro lado, el perfil de seguridad fue favorable a avalglucosidasa alfa35,38.
Otro agente nuevo es la combinación de la enzima cipaglucosidasa alfa con miglustat, que actúa como un estabilizador de la enzima. A diferencia de avalglucosidasa alfa, que está indicado para el tratamiento de todo el espectro de pacientes con enfermedad de Pompe, cipaglucosidasa alfa en combinación con miglustat está solo indicado para el tratamiento de pacientes adultos con enfermedad de Pompe de inicio tardío39–41. Esta enzima recombinante también tiene mayor afinidad por el receptor M6P independiente de cationes42. El estudio PROPEL, ensayo clínico fase III multicéntrico multinacional, mostró que los pacientes tratados con cipaglucosidasa alfa en combinación con miglustat conseguían un incremento mayor de la distancia recorrida en la prueba 6MWT (20,0 vs. 8,3m) y un menor empeoramiento de la FVC (−1,4 vs. −3,7%) a las 52 semanas de seguimiento, aunque estos resultados tampoco alcanzaron la superioridad estadística cuando se compararon con los pacientes tratados con alglucosidasa alfa40,42. En este caso se incluyeron pacientes tanto naïve (n=27) como pacientes que ya estaban en tratamiento con alglucosidasa alfa (n=95) y se aleatorizaron 2:1 a recibir cipaglucosidasa alfa y miglustat o a mantener tratamiento con alglucosidasa alfa durante 51 semanas. Al analizar los resultados según el tratamiento previo con ERT, se observó que los pacientes naïve obtenían mejores resultados en la función motora y función pulmonar con alglucosidasa alfa. La mejora de la distancia en la prueba 6MWT de los pacientes tratados con cipaglucosidasa alfa más miglustat fue de 28,5m, mientras que fue de 52,7m para los tratados con alglucosidasa alfa. Asimismo, el empeoramiento en la FVC fue mayor para los pacientes tratados con cipaglucosidasa alfa más miglustat comparado con alglucosidasa alfa (−5,2 vs. −2,4%). En cambio, los pacientes tratados previamente con ERT que cambiaron a cipaglucosidasa alfa combinada con miglustat presentaron una mayor mejora media en la distancia recorrida en la prueba 6MWT (15,9 vs. 1,0m) y un menor empeoramiento de la FVC (−0,2 vs. −3,8%) que los pacientes que mantuvieron el tratamiento con alglucosidasa alfa40. La incidencia de eventos adversos fue similar entre los 2 grupos de tratamiento, con caídas, dolor de cabeza, nasofaringitis, mialgia, artralgia y náusea como eventos más frecuentes42. Cabe destacar que la combinación cipaglucosidasa alfa más miglustat no está recomendada en mujeres en edad fértil que no utilicen métodos anticonceptivos fiables, durante el embarazo o en periodo de lactancia, debido a contraindicaciones asociadas con miglustat41.
Por tanto, las 2 nuevas terapias enzimáticas han demostrado que son por lo menos igual de eficaces y seguras que alglucosidasa alfa, al menos durante el primer año de tratamiento. Además, el tratamiento continuado con avalglucosidasa alfa ha demostrado que mantiene los beneficios a largo plazo, tal y como mostró la fase de extensión del estudio COMET. En ella a partir de la semana 49 todos los pacientes fueron tratados con avalglucosidasa alfa hasta la semana 145. Los pacientes que cambiaron de alglucosidasa alfa a avalglucosidasa alfa presentaron un cambio medio del 0,7% en la FVC (IC 95%: −1,4-2,8) y un cambio medio en la prueba 6MWT de −2,3m (IC 95%: −23,2-18,7) entre las semanas 49 y 145, mientras que aquellos con continuaron con avalglucosidasa alfa mantuvieron la mejora de la función pulmonar y motora conseguida en la semana 4939. En consecuencia, aquellos pacientes en tratamiento con alglucosidasa alfaque experimentan progresión de la enfermedad, se recomienda cambiar el tratamiento a una de las nuevas terapias de reemplazo enzimático35. No existen estudios que comparen la eficacia de las 2 nuevas terapias, por lo que no es posible recomendar una de las 2 como primera opción. Se recomienda mantener a los pacientes un mínimo de 12 meses con cada tratamiento para evaluar su eficacia antes de proponer un cambio. En casos aislados con deterioro objetivo importante se podría valorar el cambio de ERT antes de los 12 meses.
Incremento en la dosis de terapia enzimática sustitutivaLa dosis estándar de alglucosidasa alfa es de 20mg/kg, sin embargo, se ha visto que los pacientes con IOPD pueden experimentar deterioro clínico a pesar del tratamiento recomendado. Se ha demostrado que los pacientes con IOPD tratados con dosis elevadas de alglucosidasa alfa (40mg/kg/semana) mejoran la supervivencia comparados con los tratados con la dosis estándar. El aumento de la dosis también se ha asociado con una tendencia a mejorar la función motora43,44. Asimismo, también se ha demostrado el beneficio de doblar la dosis de ERT en pacientes pediátricos con LOPD. El tratamiento con ERT a dosis de 40mg/kg se ha mostrado seguro y eficaz, con mejoras en la función motora y la función pulmonar de los pacientes45.
Estos resultados apoyarían el aumento de dosis de 20 a 40mg/kg en los pacientes pediátricos que han empeorado los síntomas después de una mejora inicial con ERT, pero no existen datos suficientes para apoyar un incremento en la dosis de los pacientes adultos afectos de la enfermedad.
Estudio de anticuerpos anti-terapia enzimática sustitutivaLa formación de anticuerpos IgG contra la ERT es un efecto conocido del tratamiento. En la enfermedad de Pompe la mayoría de los pacientes en tratamiento con ERT desarrollan anticuerpos antiGAA, aunque a títulos no elevados. En los pacientes con LOPD es excepcional que estos anticuerpos neutralicen el efecto del tratamiento46, sin embargo, es recomendable su determinación en el caso de una respuesta pobre a la ERT8,47.
Tratamiento en los pacientes con afectación respiratoriaAunque la ERT mejora la calidad de vida y la supervivencia de los pacientes, la función motora y respiratoria puede continuar deteriorándose y los pacientes a menudo requieren ventilación asistida no invasiva o invasiva1,4.
Ventilación mecánica no invasivaEs necesario asegurar una ventilación diurna y nocturna adecuadas, dando soporte ventilatorio en caso necesario.
Se debe considerar iniciar la VMNI cuando hay síntomas sugestivos de hipoventilación como la somnolencia diurna, la cefalea matutina y el embotamiento matutino o síntomas sugestivos de afectación diafragmática como la ortopnea, además de hipercapnia diurna, capnografía patológica u otras pruebas funcionales patológicas (tabla 2)25,26,28.
Criterios para iniciar la ventilación mecánica25,26,28
| Indicaciones ventilación mecánica no invasiva |
| - Síntomas sugestivos de hipoventilación: somnolencia diurna, cefalea matutina, embotamiento matutino |
| - Síntomas sugestivos de afectación diafragmática: ortopnea |
| - Hipercapnia diurna |
| - TcpCO2 con pico >49mmHg y/o TcpCO2 media >50mmHg |
| - FVC<80% y/o caída >20% de la FVC en decúbito supino |
| - SNIP<40cmH2O |
| - PIM<50% y/o <−60cm de agua |
| - Pulsioximetría nocturna basal con SpO2<88%>5min consecutivos y/o SpO2<90% durante más del 10% del registro (CT90>10%) |
| Indicaciones ventilación mecánica invasiva |
| - VMNI no eficaz en corregir la hipoventilación nocturna |
| - Mal manejo de secreciones bronquiales a pesar del adecuado tratamiento |
cmH2O: centímetro de agua; CT90: porcentaje de tiempo con saturación de O2 por debajo del 90%; FVC: capacidad vital forzada; PIM: presión inspiratoria máxima; SNIP: presión inspiratoria nasal; SpO2: saturación de oxígeno; TcpCO2: presión transcutánea de dióxido de carbono; VMNI: ventilación mecánica no invasiva.
En los pacientes con LOPD con debut respiratorio en forma de fallo respiratorio agudo se debe iniciar de forma inmediata el tratamiento con soporte ventilatorio en la modalidad requerida, plantear la necesidad de adaptación a terapias de soporte respiratorio domiciliario e iniciar tratamiento con ERT.
Ventilación mecánica invasivaSe debe considerar iniciar la ventilación mecánica invasiva (VMI) cuando la VMNI no es eficaz en corregir la hipoventilación nocturna o cuando hay un mal manejo de las secreciones bronquiales a pesar del adecuado tratamiento (tabla 2)25,26,28.
Sin embargo, es necesario individualizar la decisión de iniciar la ventilación invasiva teniendo en cuenta si hay afectación de otros órganos, así como el soporte que va a necesitar el paciente.
Manejo integral del pacienteEl manejo de la enfermedad de Pompe requiere un abordaje integral y multidisciplinar del paciente, además del tratamiento específico con ERT7. Algunas medidas generales deben incluir tener una inmunización adecuada, asegurar el correcto aclaramiento de las secreciones, favorecer el mantenimiento de las capacidades funcionales del paciente y asegurar una nutrición adecuada.
La inmunización adecuada se consigue cumpliendo con el calendario completo de vacunación, además de recibir las vacunas antineumocócica y de la gripe, y mediante el tratamiento profiláctico frente al virus respiratorio sincitial6,14. Asimismo, es recomendable la vacunación frente a la COVID-19 en los pacientes con LOPD al ser población de riesgo.
Cuando hay debilidad de los músculos respiratorios la capacidad de aclaramiento de las vías respiratorias se ve reducida debido a tos ineficaz. Esto aumenta el riesgo de infecciones respiratorias y de morbimortalidad durante los procesos respiratorios agudos. Los mecanismos para mantener un drenaje adecuado de las secreciones incluyen la tos asistida manual y la tos asistida mecánica. Ante la sospecha de un mal manejo de las secreciones es recomendable medir el pico flujo de tos. Si este es menor a 270l/min se debe valorar la necesidad de tos asistida, y realizar prueba de tolerancia y efectividad de las técnicas de tos asistida tanto mecánica como manual para utilizar la más indicada26,48.
Independientemente de la afectación respiratoria es recomendable la fisioterapia respiratoria preventiva, con el objetivo de mantener la compliancia torácica y pulmonar y evitar la aparición de microatelectasias26. La fisioterapia respiratoria está dirigida a estimular el aclaramiento de las secreciones mediante la tos asistida con dispositivo (Cough Assist In-Exsufflator), maniobras de percusión (clapping), vibraciones, drenaje postural y entrenamiento de la musculatura respiratoria para mejorar la resistencia49,50. Asimismo, la rehabilitación, la terapia ocupacional y la fisioterapia favorecen el mantenimiento de las capacidades funcionales del paciente, además de mejorar los síntomas respiratorios y los trastornos del habla y la disfagia14.
Es recomendable que los pacientes realicen ejercicio físico aeróbico. Hay estudios que indican que la combinación de entrenamientos aeróbicos, de resistencia y de estabilidad de la zona central del cuerpo mejoran la resistencia, la capacidad de estabilización de los músculos axiales y la función muscular51,52. Es recomendable que los pacientes hagan ejercicio submáximo, funcional y aeróbico regular combinando ejercicios aeróbicos y de resistencia.
Finalmente, es frecuente que los pacientes con una enfermedad neuromuscular progresiva tiendan a ser menos activos físicamente y gasten menos energía que los individuos activos. Como consecuencia muchos pacientes con LOPD tienen obesidad sarcopénica, con poca masa muscular y un alto porcentaje de grasa52. Además, algunas complicaciones de la enfermedad como la disfagia pueden dificultar la ingesta de alimentos9.
Es necesario un correcto control del estado nutricional, y en caso necesario el uso de vías alternativas de alimentación cuando la oral no es suficiente o segura. La dieta debe proporcionar una cantidad adecuada de calorías y ser rica en proteínas para ayudar a prevenir la pérdida de masa muscular8,9.
| Recomendaciones para el tratamiento de la LOPD |
| • El tratamiento con ERT debe iniciarse ante los pacientes con síntomas objetivos de afectación muscular y/o respiratoria. |
| • Tras confirmar buena tolerancia a la ERT en el hospital de día se puede proponer tratamiento domiciliario. |
| • Se recomienda sustituir la ERT si se demuestra ausencia de respuesta o empeoramiento clínico progresivo. |
| • En aquellos casos pediátricos con diagnóstico de LOPD que están empeorado los síntomas tras una mejoría inicial después del tratamiento con ERT puede considerarse doblar la dosis de ERT. |
| • Se recomienda estudiar la presencia de anticuerpos anti-ERT en aquellos casos en los que no se evidencie ninguna mejoría o ante un empeoramiento clínico remarcable. |
| • Es recomendable la vacunación anual antigripal, frente al SARS-CoV-2 y frente al neumococo. |
| • La fisioterapia respiratoria en el paciente con LOPD debe recomendarse siempre, independientemente de la afectación respiratoria. |
| • Es recomendable que los pacientes realicen ejercicio físico regular. |
| Tratamiento en los pacientes con afectación respiratoria |
| • Considerar iniciar la ventilación mecánica no invasiva ante la presencia de síntomas o resultados patológicos en las pruebas funcionales e hipercapnia o capnografía patológica. |
| • Se debe considerar iniciar la ventilación mecánica invasiva cuando: |
| ∘ La VMNI no es eficaz en corregir la hipoventilación nocturna. |
| ∘ Hay un mal manejo de las secreciones bronquiales a pesar del adecuado tratamiento. |
| ∘ La indicación de ventilación mecánica invasiva debe ser individualizada. |
| • Ante la sospecha de mal manejo de secreciones por tos ineficaz, es recomendable medir el pico flujo de tos, realizar una prueba de tolerancia/efectividad de tos asistida mecánica o realizar una prueba de tolerancia/efectividad de tos asistida manual. |
El seguimiento periódico de todos los pacientes con LOPD, ya sean sintomáticos con o sin tratamiento con ERT o asintomáticos-presintomáticos, permite evaluar la progresión de la enfermedad y determinar la eficacia del tratamiento.
El seguimiento de la LOPD debe ser multidisciplinar en centros con experiencia en el manejo de la enfermedad23. Los pacientes con afectación respiratoria deben ser seguidos en consultas de neumología integradas en unidades multidisciplinares especializadas.
Se recomienda un seguimiento semestral de los pacientes sintomáticos. Las pruebas recomendadas incluyen la evaluación funcional respiratoria, la evaluación de la función motora, y el estado nutricional14,23. Los pacientes con LOPD asintomáticos deben ser evaluados al menos una vez al año para detectar precozmente el inicio de síntomas o signos de la enfermedad, en caso de producirse.
Pruebas de seguimientoEl seguimiento de los pacientes debe incluir pruebas que evalúen la fuerza muscular, la función motora, la función respiratoria y la percepción del paciente (tabla 3). Existen diferentes escalas que permiten determinar estos parámetros.
Pruebas recomendadas para el seguimiento de los pacientes
| Pruebas mínimas para el seguimiento | Otras pruebas recomendadas |
|---|---|
| - Fuerza muscular MRC | - NSAD |
| - 6MWT | - Marcha de 10m |
| - R-PAct | - Time to climb 4 steps |
| - Pruebas funcionales respiratorias | - Timpo to up&go |
MRC: Medical Research Council; NSAD: North Star Assessment for limb-girdle type muscular Dystrophies; R-Pact: Rasch-built Pompespecific Activity; 6MWT: prueba de la marcha de 6min.
La escala del Medical Research Council (MRC) se utiliza para medir la fuerza muscular. Esta escala permite medir la fuerza de diferentes grupos musculares en una escala del 0 (ausencia de movimiento) al 5 (fuerza normal)23,53.
La prueba de la marcha de 6min (6MWT) es el test cronometrado utilizado habitualmente en los ensayos clínicos. Esta prueba mide la distancia que un paciente puede caminar durante 6min en una superficie plana y dura23,54.
La escala Rasch-built Pompe-specific Activity (R-PAct) está recomendada para evaluar el impacto de la enfermedad sobre la calidad de vida. Esta escala consiste en un cuestionario con 18 preguntas que evalúan el grado de autonomía en las actividades de la vida diaria, tanto personales como sociales, y es capaz de detectar limitaciones para realizar actividades y restricciones en la participación social, así como cambios clínicamente importantes a lo largo del tiempo, en los pacientes con enfermedad de Pompe a lo largo de todo el espectro de la enfermedad23,55.
Otras pruebas que también se pueden realizar incluyen la escala de evaluación compuesta North Star Assessmentfor limb-girdle type muscular Dystrophies (NSAD), que fue diseñada específicamente para evaluar los cambios funcionales en la capacidad ambulante de pacientes con distrofia muscular por déficit de disferlina56 y se usa para pacientes adultos. La escala NSAD se ha validado en distrofias de cinturas y hay varios estudios en marcha para validarla en Pompe57. Además, se pueden usar pruebas cronometradas como la marcha de 10m, time to climb 4 steps (tiempo para subir y bajar 4 escalones) o time to up&go (tiempo para levantarse desde el suelo o de una silla)23,35.
El tetrasacárido de glucosa (Glc4) urinaria es un biomarcador cuya concentración se encuentra elevada en los pacientes con enfermedad de Pompe. Los estudios indican que los niveles de Glc4 estarían correlacionados con la edad de aparición de los síntomas y el fenotipo de la enfermedad58. Sin embargo, no es específico de la enfermedad de Pompe (niveles elevados de Glc4 pueden presentarse en infecciones urinarias, pancreatitis aguda, trauma muscular y algunos cánceres58), y los pacientes con LOPD pueden presentar niveles normales en las fases tempranas de la enfermedad20.
En la actualidad existen datos limitados de la utilidad de la Glc4 como biomarcador de gravedad o respuesta al tratamiento en los pacientes con LOPD.
Seguimiento de los pacientes con afectación pulmonarEs recomendable que las pruebas funcionales respiratorias se realicen al menos cada 6 a 8 meses, tanto en los pacientes ventilados como no ventilados, para hacer un seguimiento adecuado de la progresión de la enfermedad y determinar la necesidad de ventilación o de aumentar las horas de ventilación25,26.
En los pacientes no ventilados se debe realizar una oxi-capnografía nocturna basal para detectar si hay periodos de hipoventilación nocturna.
Los pacientes con ventilación, además, pueden disponer de los datos del built-in-software del ventilador, que informan del tiempo de uso del mismo, el volumen corriente, la frecuencia respiratoria, el número de respiraciones activada por el paciente o las fugas, y permiten conocer la eficacia de la VMNI59. En función de los resultados también se puede realizar una oxi-capnografía y un estudio del sueño (en aquellos pacientes con ventilación no invasiva)23,35.
| Recomendaciones para el seguimiento de la LOPD |
| • Es recomendable que en el seguimiento de los pacientes estén involucradas las especialidades de neurología/neuropediatría/medicina interna, neumología, rehabilitación y nutrición. |
| • Los pacientes asintomáticos deben evaluarse al menos una vez al año, mientras que los pacientes sintomáticos deben evaluarse al menos cada 6 meses. |
| • Para el seguimiento de los pacientes están especialmente recomendadas la escala manual para medir fuerza muscular, 6MWT, escala R-PAcT y pruebas funcionales respiratorias; y si es posible se recomienda realizar la NSAD y alguna otra prueba cronometrada como tiempo de caminar 10m, time to up&go y/o time to climb 4 steps. |
| • No hay datos suficientes para recomendar el uso de tetrasacárido de glucosa (Glc4) para el seguimiento de la LOPD. |
| Seguimiento en los pacientes con afectación respiratoria |
| • Los pacientes con afectación respiratoria deben ser seguidos en consultas de neumología integradas en unidades multidisciplinares especializadas. |
| • Los pacientes deben ser evaluados por neumología cada 6 a 8 meses. |
| • Las pruebas recomendadas para el seguimiento del paciente con afectación respiratoria incluyen: |
| ∘ En los pacientes no ventilados: gasometría, FVC, PIM y PEM, SNIP, pico flujo de tos y oxi-capnografía. |
| ∘ En los pacientes ventilados: gasometría, FVC, PIM y PEM, SNIP, pico flujo de tos, built-in-software y, en función de los resultados, oxi-capnografía y estudio de sueño. |
| ∘ Ecografía diafragmática y/o RM como pruebas opcionales. |
La LOPD es una enfermedad progresiva que puede llevar a un deterioro funcional importante del paciente, con una gran dependencia en las actividades de la vida diaria, así como un estado de insuficiencia respiratoria avanzado. En esta fase de la enfermedad, los tratamientos están dirigidos a manejar los síntomas para mantener la mejor calidad de vida posible y dar apoyo al paciente y los familiares hasta el final de la vida52.
Para la toma de decisiones finales es aconsejable que se elabore un documento de voluntades anticipadas, en el que se recogen las decisiones del paciente sobre aquellos cuidados y tratamientos que desea recibir o no cuando su situación no le permita expresar su voluntad. Las guías SEPAR 2012 proporcionan unas pautas a seguir para el manejo del paciente con enfermedad neuromuscular en el final de la vida26.
FinanciaciónSanofi financió la reunión de expertos. Los autores no recibieron ningún pago de Sanofi en relación con el desarrollo de esta publicación. Sanofi tuvo la oportunidad de revisar la publicación; Sin embargo, los autores siguen siendo responsables de todo el contenido, las decisiones editoriales y la decisión de enviar el manuscrito.
Conflicto de interesesLos autores declararon no tener conflictos de intereses, excepto:
Cristina Domínguez-González ha recibido honorarios de Sanofi, Amicus Therapeutics y Astellas Pharma por la realización de servicios de consultoría y ponencias.
Jordi Díaz Manera ha recibido fondos para proyectos de investigación de Sarepta, Sanofi, Spark, Amicus, Astellas y Boehringer-Ingelheim y ha recibido pagos por participar en Advisory Boards o presentar datos representando a Amicus, Astellas, Spark, Sarepta, Lupin y Sanofi.
Javier de las Heras ha recibido honorarios por parte de Sanofi por participación en eventos de formación médica y consultoría.
Raúl Juntas Morales declara haber recibido compensación económica de parte de Sanofi por la participación en advisory boards y realización de conferencias.
Juan Carlos León Hernández declara haber recibido honorarios por ponencias y moderación de encuentros médicos a cago de Sanofi-Genzyme.
Nuria Muelas declara haber participado como asesora de Sanofi-Genzyme, Amicus y Astellas Therapeutics y recibido remuneraciones por impartir charlas formativas.
Montse Olivé declara haber recibido honorarios de Sanofi y Amicus Therapeutics en concepto de consultoría o participación como ponente en eventos científicos.
Cristina Domínguez-González, Montse Olivé, Carmen Paradas, Nuria Muelas, Andrés Nascimento y Raúl Juntas forman parte de la Red Europea de Centros de Referencia, ERN-NMD.

