




La neuropatía hereditaria con parálisis sensible a la presión (NHPP) es una enfermedad inusual de la mielina periférica, que se hereda de forma autosómica dominante, y se manifiesta mediante neuropatías recurrentes sensoriales y/o motoras indoloras y, normalmente, transitorias1–3. Las neuropatías son desencadenadas por traumatismos menores o movimientos repetitivos con compresión y tracción de los nervios periféricos. Las formas de presentación más habituales se producen en el nervio peroneo y en el nervio cubital. La plexopatía braquial tiene lugar en un 11-20% de los casos a lo largo del curso natural de la enfermedad, pero es inusual como manifestación clínica, y más aún si se presenta bilateralmente1,2. A continuación presentamos el caso clínico de un paciente con NHPP manifestada inusualmente en forma de plexopatía braquial bilateral.
Un varón de 22 años de edad con historial personal y familiar ordinaria fue transferido a nuestro departamento al sufrir debilidad en la musculatura proximal del miembro superior izquierdo durante un periodo de 3 semanas. El paciente notó la debilidad al despertarse y no sentía dolor, no tenía historial de traumatismos, ni antecedentes de intervenciones quirúrgicas, ni infecciones. Curiosamente 4 meses antes, el paciente había presentado una sintomatología similar en el miembro superior derecho con debilidad en la musculatura proximal, indolora y parestesia en la mano, de la que aún se estaba recuperando y que también notó al despertarse. En esta primera fase, se llevó a cabo un estudio de electroneuromiografía (EMG) (tablas 1 y 2) que constataba una plexopatía braquial derecha, ampliamente extendida en el tronco superior y latencia distal ligeramente prolongada de los nervios mediano motor, mediano sensitivo y cubital sensitivo derechos. Durante la segunda fase, fue observado en nuestro departamento, y en la exploración se objetivaba una fuerza muscular 4/5 en la abducción y rotación externa del hombro derecho y una fuerza muscular 3/5 en la abducción y 4/5 en la rotación externa del hombro izquierdo, los restantes segmentos musculares no presentaban paresia; los reflejos osteotendinosos eran débiles en los miembros superiores; la restante exploración no tenía alteraciones. Se practicó una EMG (tablas 1 y 2) en nuestro departamento, la cual mostró una plexopatía braquial izquierda con una lesión mayor del tronco superior, así como una neuropatía del nervio cubital izquierdo en el codo sin otras alteraciones. La resonancia magnética (RM) cervical fue normal y la RM de alta resolución de los plexos braquiales (fig. 1) mostró un aumento de la señal en los músculos supraespinoso, infraespinoso y redondo menor izquierdos, lo que apuntaba a una denervación muscular carente de lesión estructural del plexo braquial izquierdo. Los análisis de sangre ofrecieron resultados normales, incluyendo la autoinmunidad. Al apuntar tanto las manifestaciones clínicas como los resultados neurofisiológicos a un diagnóstico de NHPP, se solicitó un test genético que reveló una deleción de heterocigoto (17p11.2-12) en el gen de la proteína mielina periférica (PMP22) y confirmó el diagnóstico. El paciente recuperó la fuerza en la musculatura de los miembros superiores con la rehabilitación, y se adoptaron medidas preventivas.
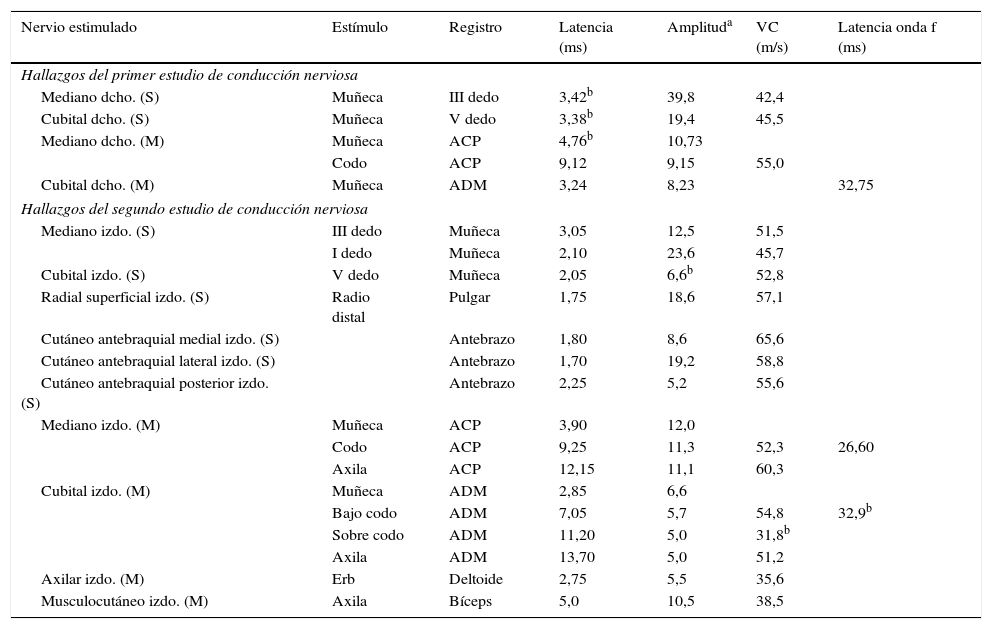
Resultados de los estudios de conducción nerviosa
| Nervio estimulado | Estímulo | Registro | Latencia (ms) | Amplituda | VC (m/s) | Latencia onda f (ms) |
|---|---|---|---|---|---|---|
| Hallazgos del primer estudio de conducción nerviosa | ||||||
| Mediano dcho. (S) | Muñeca | III dedo | 3,42b | 39,8 | 42,4 | |
| Cubital dcho. (S) | Muñeca | V dedo | 3,38b | 19,4 | 45,5 | |
| Mediano dcho. (M) | Muñeca | ACP | 4,76b | 10,73 | ||
| Codo | ACP | 9,12 | 9,15 | 55,0 | ||
| Cubital dcho. (M) | Muñeca | ADM | 3,24 | 8,23 | 32,75 | |
| Hallazgos del segundo estudio de conducción nerviosa | ||||||
| Mediano izdo. (S) | III dedo | Muñeca | 3,05 | 12,5 | 51,5 | |
| I dedo | Muñeca | 2,10 | 23,6 | 45,7 | ||
| Cubital izdo. (S) | V dedo | Muñeca | 2,05 | 6,6b | 52,8 | |
| Radial superficial izdo. (S) | Radio distal | Pulgar | 1,75 | 18,6 | 57,1 | |
| Cutáneo antebraquial medial izdo. (S) | Antebrazo | 1,80 | 8,6 | 65,6 | ||
| Cutáneo antebraquial lateral izdo. (S) | Antebrazo | 1,70 | 19,2 | 58,8 | ||
| Cutáneo antebraquial posterior izdo. (S) | Antebrazo | 2,25 | 5,2 | 55,6 | ||
| Mediano izdo. (M) | Muñeca | ACP | 3,90 | 12,0 | ||
| Codo | ACP | 9,25 | 11,3 | 52,3 | 26,60 | |
| Axila | ACP | 12,15 | 11,1 | 60,3 | ||
| Cubital izdo. (M) | Muñeca | ADM | 2,85 | 6,6 | ||
| Bajo codo | ADM | 7,05 | 5,7 | 54,8 | 32,9b | |
| Sobre codo | ADM | 11,20 | 5,0 | 31,8b | ||
| Axila | ADM | 13,70 | 5,0 | 51,2 | ||
| Axilar izdo. (M) | Erb | Deltoide | 2,75 | 5,5 | 35,6 | |
| Musculocutáneo izdo. (M) | Axila | Bíceps | 5,0 | 10,5 | 38,5 | |
ACP: abductor corto del pulgar; ADM: abductor del dedo meñique; dcho.: derecho; izdo.: izquierdo; M: motor; S: sensitivo; VC: velocidad de conducción.
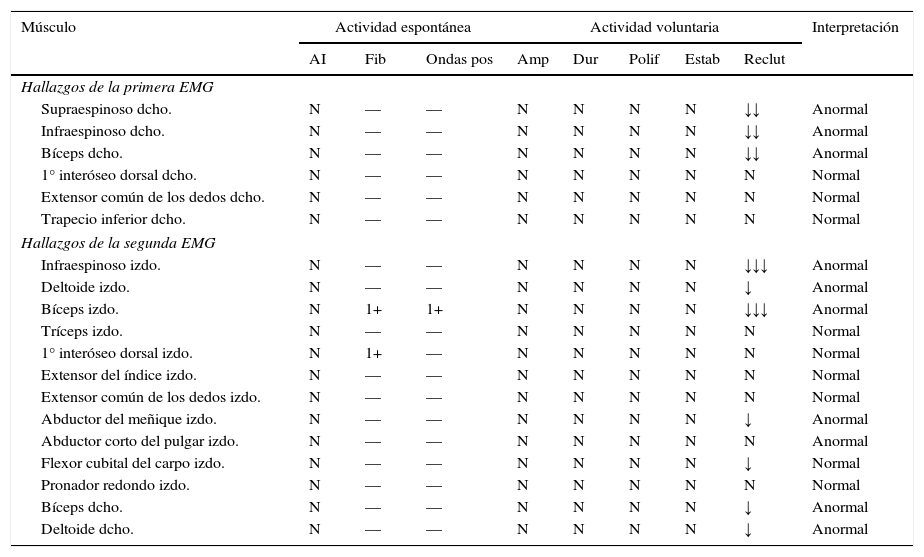
Resultados de las EMG
| Músculo | Actividad espontánea | Actividad voluntaria | Interpretación | ||||||
|---|---|---|---|---|---|---|---|---|---|
| AI | Fib | Ondas pos | Amp | Dur | Polif | Estab | Reclut | ||
| Hallazgos de la primera EMG | |||||||||
| Supraespinoso dcho. | N | — | — | N | N | N | N | ↓↓ | Anormal |
| Infraespinoso dcho. | N | — | — | N | N | N | N | ↓↓ | Anormal |
| Bíceps dcho. | N | — | — | N | N | N | N | ↓↓ | Anormal |
| 1° interóseo dorsal dcho. | N | — | — | N | N | N | N | N | Normal |
| Extensor común de los dedos dcho. | N | — | — | N | N | N | N | N | Normal |
| Trapecio inferior dcho. | N | — | — | N | N | N | N | N | Normal |
| Hallazgos de la segunda EMG | |||||||||
| Infraespinoso izdo. | N | — | — | N | N | N | N | ↓↓↓ | Anormal |
| Deltoide izdo. | N | — | — | N | N | N | N | ↓ | Anormal |
| Bíceps izdo. | N | 1+ | 1+ | N | N | N | N | ↓↓↓ | Anormal |
| Tríceps izdo. | N | — | — | N | N | N | N | N | Normal |
| 1° interóseo dorsal izdo. | N | 1+ | — | N | N | N | N | N | Normal |
| Extensor del índice izdo. | N | — | — | N | N | N | N | N | Normal |
| Extensor común de los dedos izdo. | N | — | — | N | N | N | N | N | Normal |
| Abductor del meñique izdo. | N | — | — | N | N | N | N | ↓ | Anormal |
| Abductor corto del pulgar izdo. | N | — | — | N | N | N | N | N | Anormal |
| Flexor cubital del carpo izdo. | N | — | — | N | N | N | N | ↓ | Normal |
| Pronador redondo izdo. | N | — | — | N | N | N | N | N | Normal |
| Bíceps dcho. | N | — | — | N | N | N | N | ↓ | Anormal |
| Deltoide dcho. | N | — | — | N | N | N | N | ↓ | Anormal |
AI: actividad de inserción; Amp: amplitud; dcho.: derecho; Dur: duración; EMG: electromiografía; Estab: estabilidad; Fib: fibrilación; izdo.: izquierdo; Polif: polifasia; pos: positivas; Reclut: reclutamiento.
 con secuencias neurográficas: imagen STIR (secuencia inversión-recuperación con tiempo de inversión corto) sagital (A) mostrando un aumento de la señal (flechas) en los músculos supraespinoso, infraespinoso y redondo menor izquierdos; imagen STIR coronal (B) sin alteraciones morfológicas en los plexos braquiales.")
RM de alta resolución (3 tesla) con secuencias neurográficas: imagen STIR (secuencia inversión-recuperación con tiempo de inversión corto) sagital (A) mostrando un aumento de la señal (flechas) en los músculos supraespinoso, infraespinoso y redondo menor izquierdos; imagen STIR coronal (B) sin alteraciones morfológicas en los plexos braquiales.
Normalmente, la presentación de la NHPP se produce en la segunda o tercera década de vida, aunque la edad de inicio puede ser muy variable3,4. El diagnóstico conlleva un historial clínico de neuropatías recurrentes, un historial familiar constante de herencia autosómica dominante, investigaciones sobre conducción nerviosa con neuropatías sensitivo-motoras focales desmielinizantes en sitios de atrapamiento y/o una polineuropatía difusa, un engrosamiento mielínico focal en la biopsia nerviosa cuando fuere justificable y, por último, la confirmación del test genético1–4. La mayoría de los pacientes presentan deleción en el cromosoma 17p11.2 incluyendo el gen PMP22, y los restantes sufren mutación en el gen en sí5,6.
Se observa gran heterogeneidad fenotípica en la NHPP, incluso dentro de la misma familia. La plexopatía braquial puede darse en algunos pacientes, pero generalmente se asocia a otras mononeuropatías recurrentes a lo largo del transcurso de la enfermedad1,2. La corta duración de la enfermedad en nuestro paciente puede explicar la ausencia de sintomatología debido a la actuación de otro nervio, y el historial familiar negativo puede deberse a una enfermedad asintomática en uno de sus progenitores o a una supresión de novo en nuestro paciente. Pocas de las familias, con NHPP cuyos casos se han publicado, tienen plexopatías braquiales recurrentes como única manifestación clínica7–12, y en la mayoría de esas familias los síntomas comenzaron al despertarse, lo que sugiere una mayor vulnerabilidad del plexo braquial a los factores mecánicos durante el sueño11.
Más allá de la NHPP, el diagnóstico diferencial de plexopatía braquial no traumática debe incluir lesiones compresivas, la amiotrofia neurálgica hereditaria y la amiotrofia neurálgica idiopática (síndrome de Parsonage-Turner), pero en estas enfermedades la dolor es una de las manifestaciones principales y las 2 últimas se asocian característicamente a daño axonal localizado al plexo en los estudios neurofisiológicos10,11.
La NHPP debería considerarse al evaluar a pacientes con plexopatía braquial indolora no traumática principalmente desmielinizante. Hasta donde sabemos, nunca se había publicado sobre la NHPP que se presenta como plexopatía braquial bilateral.
FinanciaciónNuestro trabajo ha sido redactado sin financiación por ninguna empresa o entidad pública o privada.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses, y se ha obtenido el consentimiento informado del paciente a autorizar la publicación de su caso.

