



La neuropatía hereditaria con parálisis sensible a la presión (NHPP) es una alteración autosómica dominante, con episodios recurrentes de mononeuropatía en nervios susceptibles de compresión típicos, con características neurofisiológicas comunes. Sin embargo, se han comunicado otras presentaciones clínicas y neurofisiológicas.
MétodosAnálisis de características clínicas y neurofisiológicas en una revisión retrospectiva de 20 pacientes con NHPP confirmados genéticamente, de los cuales, 16 se estudiaron en nuestro servicio entre los años 1996 y 2016.
ResultadosAdemás de las características típicas de la NHPP, encontramos formas atípicas como síntomas sensitivos recurrentes posicionales en 3 pacientes, polineuropatía sensitivo motora crónica en uno y mononeuropatía no evolutiva en otro. Dos pacientes empezaron en edad temprana, uno a los 7 años, con lesión del ciático poplíteo externo y otro al nacer, con afectación de plexo braquial. Las principales alteraciones de la conducción nerviosa que se encontraron fueron: la disminución de la velocidad de conducción sensitiva con rangos del 84% en el sural y 94% en el mediano y peroneo superficial, descenso de la velocidad de conducción motora del nervio cubital a través del codo en el 97% y el incremento de la latencia distal motora del nervio mediano y ciático poplíteo externo en el 74%.
ConclusiónDe acuerdo con los resultados, confirmamos la variabilidad clínica de la NHPP y encontramos los hallazgos más frecuentes en la conducción nerviosa, la disminución generalizada de la velocidad de conducción sensitiva, además de la afectación motora principalmente en lugares susceptibles de compresión, y detectamos los casos típicos, atípicos y asintomáticos de esta en entidad.
Hereditary neuropathy with liability to pressure palsy (HNPP) is an autosomal dominant disorder, typically presenting with recurrent episodes of mononeuropathy in nerves susceptible to compression, with similar neurophysiological characteristics. However, other clinical and neurophysiological presentations have been reported.
MethodsWe retrospectively analysed the clinical and neurophysiological characteristics of 20 patients with genetically confirmed HNPP. Sixteen patients were studied in our department between 1996 and 2016.
ResultsIn addition to the typical characteristics of HNPP, we found atypical forms including recurrent positional sensory symptoms in 3 patients, chronic sensorimotor polyneuropathy in one, and non-progressive mononeuropathy in one. Onset was early in 2 patients: one at the age of 7 years, with common peroneal nerve injury, and another at birth, with brachial plexus involvement. By frequency, the main pathological findings in the nerve conduction study were: decreased sensory nerve conduction velocity in the sural (84%) and the median and superficial peroneal nerves (94%); decreased motor nerve conduction velocity in the ulnar nerve through the elbow (97%), and increased motor distal latency of the median and deep peroneal nerves (74%).
ConclusionOur results confirm the clinical variability of HNPP, with the most frequent nerve conduction study findings being the generalised decrease in sensory nerve conduction velocity, in addition to motor involvement, mainly in locations susceptible to nerve compression. The nerve conduction study can detect typical, atypical, and asymptomatic cases of HNPP.
La neuropatía hereditaria sensible a la presión (NHPP) es una neuropatía periférica que comienza alrededor de la segunda o tercera décadas de la vida. Hay pocos casos descritos en la literatura de inicio temprano. Típicamente presenta episodios agudos recurrentes de parálisis de nervios sin dolor, en lugares anatómicos susceptibles de atrapamiento, precedidos por un traumatismo menor o compresión, con una recuperación que en la mayoría de los casos es completa. Se han descrito presentaciones clínicas atípicas como son la mononeuropatía no evolutiva1,2, polineuropatía sensitivo-motora (PNP S-M) crónica, mononeuropatía progresiva sensitivo-motora3, neuropatía por atrapamiento multifocal, PNP progresiva crónica Charcot-Marrie-Tooth like4, parálisis radial aguda bilateral, síntomas sensitivos recurrentes a corto plazo y síndrome escapuloperoneal5.
Su mecanismo fisiopatológico exacto es desconocido. La mayoría de los casos se asocian a una deleción de 1,5 Mb en la región 17p11.2 que contiene el gen de la peripheral myelin protein 22 (PMP 22)6-8. De forma menos frecuente se pueden encontrar mutaciones de dicho gen4.
La existencia de casos asintomáticos9-12 y su curso benigno2,3 hacen que sea una enfermedad infradiagnosticada. El estudio neurofisiológico permite detectar, además de a los pacientes con clínica típica y atípica13-21, a los familiares asintomáticos, y con ello, identificar a los portadores del gen2,19. Concierne conocer mejor las características neurofisiológicas de la NHPP. Los hallazgos más frecuentes se observan en la conducción motora4,12,16,22,23, principalmente en los sitios susceptibles de compresión e incluyen: aumento de la latencia distal motora (LDM) del nervio mediano, disminución de la velocidad de conducción motora (VCM) a través del codo del nervio cubital y un aumento de la LDM o disminución de la VCM en al menos uno de los nervios ciático poplíteo externo (CPE). Con menor frecuencia se han descrito alteraciones de la conducción nerviosa sensitiva22,24,25, como la disminución de la amplitud sensitiva en las extremidades superiores o disminución generalizada de la velocidad de conducción sensitiva (VCS). Además, se ha descrito un patrón de afectación constante que consiste en la disminución de la VCS en el segmento palma-muñeca de ambos nervios medianos3,13.
Debido a esta variabilidad de presentaciones, nuestro objetivo es analizar las características neurofisiológicas y clínicas de los pacientes con deleción 17p11.2 vistos en nuestro centro en un periodo de 20 años.
Material y métodosSe realizó un análisis retrospectivo de la historia de 20 pacientes con deleción 17p11.2 del gen PMP22, valorados en el Complejo Hospitalario de Navarra entre los años 1996 y 2016. El estudio se ha realizado de acuerdo con las normas del Comité Ético de nuestro centro.
Se recogieron datos de la historia clínica, como los antecedentes familiares de trasmisión directa o casos aislados, la edad de inicio de los síntomas, las manifestaciones clínicas, los nervios clínicamente afectados, exploración física y evolución.
El estudio neurofisiológico fue realizado en 16 pacientes, usando los protocolos estandarizados26,27 con un electromiógrafo Synergy de 5 canales, con software versión 15.0 de la empresa Natus Inc. La temperatura de la piel durante los estudios fue mantenida por encima de los 31°C. La estimulación y el registro fueron realizados con electrodos de superficie.
La conducción nerviosa motora fue estudiada en los nervios mediano, cubital, CPE y tibial posterior, y registrada en los músculos abductor corto del pulgar, abductor del meñique, extensor corto de los dedos y abductor del dedo gordo, respectivamente. La LDM se obtuvo con estimulación a una distancia de 3cm para los nervios mediano y cubital, de 5cm para el nervio CPE y de 8cm para el nervio tibial posterior. La VCM fue medida en los siguientes segmentos: muñeca a codo para el nervio mediano, muñeca a sobre codo para el nervio cubital y tobillo a fosa poplítea para los nervios CPE y tibial posterior. Se estudiaron segmentos adicionales en sitios susceptibles de compresión, en el nervio cubital a través del codo y en el nervio CPE a través de la cabeza del peroné. Se consideró alteración de la conducción en estos segmentos la disminución de 10 m/s o más de la VCM y se consideró bloqueo de conducción motora la disminución de la amplitud del potencial de acción muscular compuesto del 30%, comparando los valores proximal y distal.
En la conducción nerviosa sensitiva las respuestas se obtuvieron ortodrómicamente en extremidades superiores en el nervio mediano (de falange proximal del segundo dedo a muñeca) y en el nervio cubital (de falange proximal del quinto dedo a muñeca). En extremidades inferiores se obtuvieron antidrómicamente en el nervio sural (de mitad de la pantorrilla a maléolo lateral) y en el nervio peroneo superficial (de tercio distal lateral de la pierna al punto medio entre el maléolo lateral y el tendón del tibial anterior). La estimulación nerviosa sensitiva se hizo a una distancia de 14cm en el nervio sural y a 10cm en el nervio peroneo superficial, con una VCS medida al pico de la amplitud.
Los valores normales de referencia usados en nuestro laboratorio son los propios, y los de otros centros26-28 extendidos a nuestra población.
ResultadosSujetos y historia familiarDisponemos de datos de 20 pacientes, todos ellos con la deleción 17p11.2; 11 de sexo femenino y 9 de sexo masculino. La edad de inicio de los síntomas estuvo entre los 0 y 74 años. El 90% (18/20) pertenecían a 7 familias con historia de síntomas de NHPP, con distribución autosómica dominante. Solo un 10% (2/20) se presentaron como casos aislados, aparentemente, sin familiares afectos.
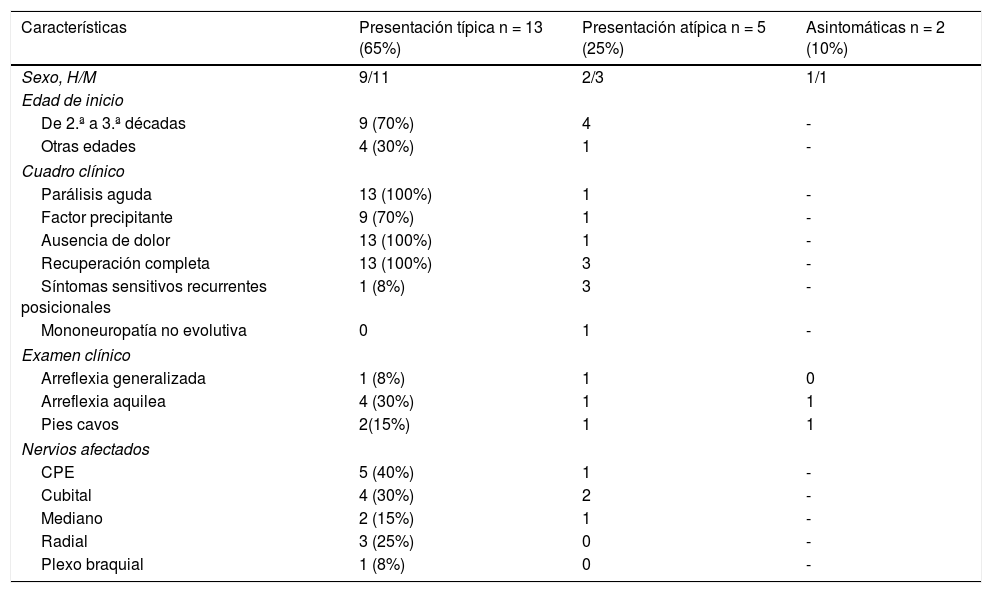
Presentación y características clínicasSe diagnosticaron 2 pacientes asintomáticos (10%) en el estudio ampliado a familiares. Del total, 13 pacientes (65%) empezaron con características clínicas consideradas como típicas y 5 pacientes (25%) con características clínicas descritas como atípicas (ver tabla 1).
Características clínicas en las presentaciones típicas, atípicas y asintomáticas
| Características | Presentación típica n = 13 (65%) | Presentación atípica n = 5 (25%) | Asintomáticas n = 2 (10%) |
|---|---|---|---|
| Sexo, H/M | 9/11 | 2/3 | 1/1 |
| Edad de inicio | |||
| De 2.ª a 3.ª décadas | 9 (70%) | 4 | - |
| Otras edades | 4 (30%) | 1 | - |
| Cuadro clínico | |||
| Parálisis aguda | 13 (100%) | 1 | - |
| Factor precipitante | 9 (70%) | 1 | - |
| Ausencia de dolor | 13 (100%) | 1 | - |
| Recuperación completa | 13 (100%) | 3 | - |
| Síntomas sensitivos recurrentes posicionales | 1 (8%) | 3 | - |
| Mononeuropatía no evolutiva | 0 | 1 | - |
| Examen clínico | |||
| Arreflexia generalizada | 1 (8%) | 1 | 0 |
| Arreflexia aquilea | 4 (30%) | 1 | 1 |
| Pies cavos | 2(15%) | 1 | 1 |
| Nervios afectados | |||
| CPE | 5 (40%) | 1 | - |
| Cubital | 4 (30%) | 2 | - |
| Mediano | 2 (15%) | 1 | - |
| Radial | 3 (25%) | 0 | - |
| Plexo braquial | 1 (8%) | 0 | - |
Los 13 pacientes con cuadro clínico típico presentaron parálisis aguda recurrente y regresiva precedida por un factor precipitante con recuperación en días o meses, con una frecuencia de nervios afectados del 40% para el CPE, 30% para el cubital, 25% para el radial, 15% para el mediano y un 8% para el plexo braquial. De los 13, 9 iniciaron los síntomas entre la 2.ª y 3.ª décadas de la vida. De ellos, 2 lo hicieron posteriormente y otros 2 iniciaron los síntomas a edades muy tempranas. Uno al nacimiento, tras el trauma del parto (fórceps), presentó datos de plexopatía braquial por debilidad a nivel proximal en extremidad superior derecha, que recuperó completamente en unos meses con tratamiento rehabilitador. Como síntomas asociados presentó hipotonía generalizada y discapacidad cognitiva. En el estudio familiar se encuentra al padre, 2 tíos paternos y un primo afectos de NHPP. Se confirma el diagnóstico por análisis genético y en el estudio neurofisiológico al año de edad. Hasta la fecha de la revisión con 15 años de edad, no ha vuelto a presentar episodios de parálisis. El segundo paciente que presentó inicio precoz mostró los síntomas a los 7 años con pies cavos y una debilidad del CPE derecho que recuperó en unos días. Como antecedentes familiares destaca que la madre, 3 tíos maternos y la abuela materna están afectos de NHPP. Actualmente, con 23 años, no ha vuelto a presentar episodios de parálisis; fue intervenido de pies cavos a los 20 años.
Respecto a los 5 pacientes con cuadros clínicos atípicos, todos ellos presentaban historia familiar de NHPP, las edades de inicio fueron entre los 24 y 60 años, con un 80% de ellos entre la 2.ª y la 3.ª décadas. Tres pacientes presentaron síntomas sensitivos recurrentes a corto plazo producidos por posturas en territorios de los nervios mediano y cubital. En otro paciente se manifestó como una polineuropatía sensitivo motora crónica que consultó a los 60 años por parestesias generalizadas crónicas y debilidad, con datos a la exploración de arreflexia generalizada, pies cavos y atrofia peroneal. Como antecedentes familiares, 2hijos de la paciente estaban diagnosticados de NHPP. El último paciente con síntomas atípicos empezó a los 24 años con cuadro clásico de múltiples parálisis en extremidades superiores e inferiores que afectaban a los nervios CPE y cubitales, durante el sueño y con relación a posturas, que se recuperaron completamente en días, con reapariciones cada uno o 2 años. A la edad de 31 años se produjo el episodio atípico por parálisis del CPE izquierdo que no recuperó y que evolucionó a una mononeuropatía crónica no evolutiva. Actualmente persiste la debilidad para la dorsiflexión del pie. Aunque inicialmente los estudios de conducción nerviosa mostraban datos de NHPP, el estudio genético no fue concluyente y a la edad de 65 años se confirmó genéticamente.
Estudio neurofisiológicoLos datos del estudio neurofisiológico corresponden a 16 de los 20 pacientes, 12 con cuadro clínico típico, 2 con cuadro clínico atípico y 2 asintomáticos (tablas 2 y 3). Las edades a las que se realizaron los registros fueron entre los 14 meses y 76 años.
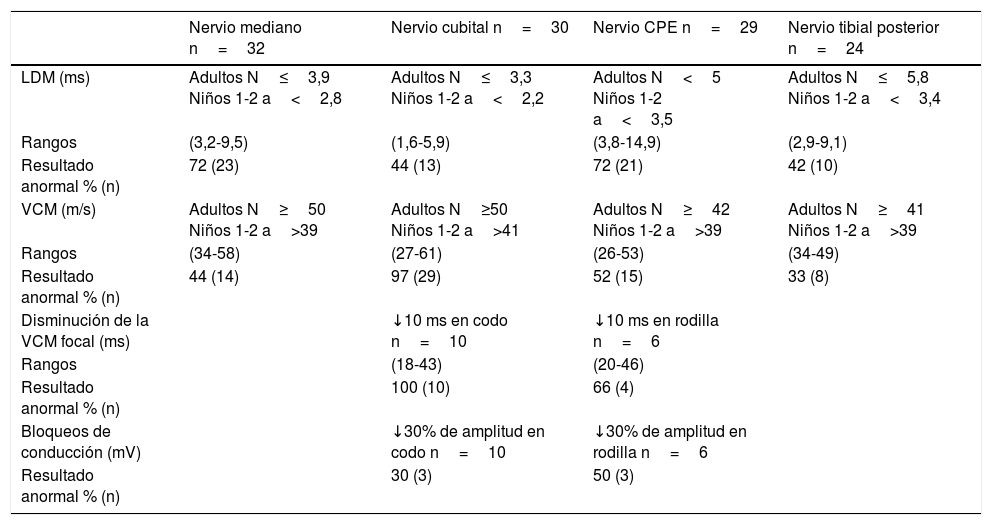
Estudio neurofisiológico en 16 pacientes con deleción 1,5Mb en el cromosoma 17p11.2: Parámetros de conducción motora
| Nervio mediano n=32 | Nervio cubital n=30 | Nervio CPE n=29 | Nervio tibial posterior n=24 | |
|---|---|---|---|---|
| LDM (ms) | Adultos N≤3,9 Niños 1-2 a<2,8 | Adultos N≤3,3 Niños 1-2 a<2,2 | Adultos N<5 Niños 1-2 a<3,5 | Adultos N≤5,8 Niños 1-2 a<3,4 |
| Rangos | (3,2-9,5) | (1,6-5,9) | (3,8-14,9) | (2,9-9,1) |
| Resultado anormal % (n) | 72 (23) | 44 (13) | 72 (21) | 42 (10) |
| VCM (m/s) | Adultos N≥50 Niños 1-2 a>39 | Adultos N≥50 Niños 1-2 a>41 | Adultos N≥42 Niños 1-2 a>39 | Adultos N≥41 Niños 1-2 a>39 |
| Rangos | (34-58) | (27-61) | (26-53) | (34-49) |
| Resultado anormal % (n) | 44 (14) | 97 (29) | 52 (15) | 33 (8) |
| Disminución de la VCM focal (ms) | ↓10 ms en codo n=10 | ↓10 ms en rodilla n=6 | ||
| Rangos | (18-43) | (20-46) | ||
| Resultado anormal % (n) | 100 (10) | 66 (4) | ||
| Bloqueos de conducción (mV) | ↓30% de amplitud en codo n=10 | ↓30% de amplitud en rodilla n=6 | ||
| Resultado anormal % (n) | 30 (3) | 50 (3) |
LDM: latencia distal motora; n: número de nervios registrados; N: valores normales para el laboratorio; VCM: velocidad de conducción motora.
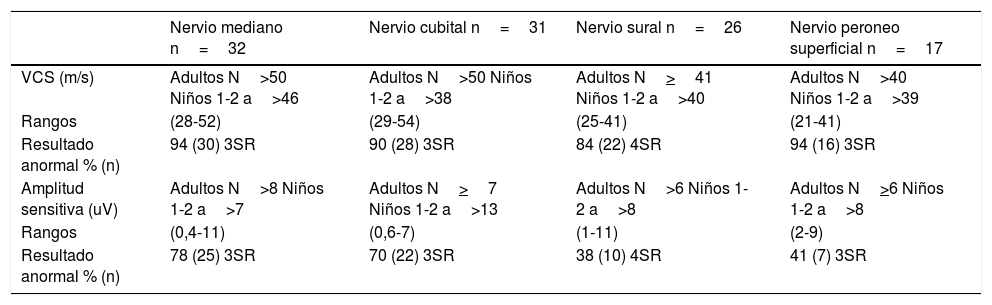
Estudio neurofisiológico en 16 pacientes con deleción 1,5Mb en el cromosoma 17p11.2: Parámetros de conducción sensitiva
| Nervio mediano n=32 | Nervio cubital n=31 | Nervio sural n=26 | Nervio peroneo superficial n=17 | |
|---|---|---|---|---|
| VCS (m/s) | Adultos N>50 Niños 1-2 a>46 | Adultos N>50 Niños 1-2 a>38 | Adultos N>41 Niños 1-2 a>40 | Adultos N>40 Niños 1-2 a>39 |
| Rangos | (28-52) | (29-54) | (25-41) | (21-41) |
| Resultado anormal % (n) | 94 (30) 3SR | 90 (28) 3SR | 84 (22) 4SR | 94 (16) 3SR |
| Amplitud sensitiva (uV) | Adultos N>8 Niños 1-2 a>7 | Adultos N>7 Niños 1-2 a>13 | Adultos N>6 Niños 1-2 a>8 | Adultos N>6 Niños 1-2 a>8 |
| Rangos | (0,4-11) | (0,6-7) | (1-11) | (2-9) |
| Resultado anormal % (n) | 78 (25) 3SR | 70 (22) 3SR | 38 (10) 4SR | 41 (7) 3SR |
n: número de nervios registrados; N: valores normales para el laboratorio; SR: nervios sin respuesta, VCS: velocidad de conducción sensitiva.
En cuanto a la conducción nerviosa sensitiva, en nuestra serie destaca el porcentaje alto de afectación de la VCS en todos los nervios estudiados, con cifras que van desde el 84% (sural) hasta el 94% (mediano y peroneo superficial). Respecto a la amplitud sensitiva, se encuentra reducida en mediano en un 78% cubital en un 70%, peroneo superficial en un 41% y sural en un 38%.
De los parámetros de la conducción sensitiva motora, la anomalía más frecuentemente encontrada fue la disminución de la VCM en el nervio cubital a través del codo (97%), le sigue el aumento de la LDM del mediano y CPE (72% cada uno). El resto de los hallazgos fueron disminución de la VCM en CPE (52%), mediano (44%) y tibial posterior (33%) y aumento de la LDM en cubital (44%) y tibial posterior (42%).
La conducción por segmentos solo se estudió en 6 pacientes para el nervio cubital a través del codo y en 3 pacientes para el nervio CPE a través de la cabeza del peroné. La disminución de la conducción en estos segmentos se observó en un 100% para el nervio cubital y en un 66% para el nervio CPE, con un 30% de nervios cubitales y un 50% de CPE con bloqueos de conducción.
DiscusiónQueremos destacar las presentaciones a edades poco frecuentes en 2pacientes de nuestra serie con antecedentes familiares de NHPP, que empezaron a edades muy tempranas, uno con parálisis del CPE a los 7 años y otro al nacimiento, con hipotonía y plexopatía braquial por el parto. Ambos evolucionaron satisfactoriamente sin volver a presentar nuevos episodios de parálisis hasta la fecha, y se diagnosticaron de forma temprana con el estudio neurofisiológico y genético. En la revisión bibliográfica que hemos realizado solo hemos hallado 3casos de inicio al nacimiento. Uno, al igual que nuestro paciente, con clínica de una parálisis transitoria de Erb8, el segundo con un CPE y pies cavos9 y un tercero que presentó al nacimiento una parálisis del CPE con recuperación completa y que se diagnosticó tardíamente, a los 7 años, por debilidad muscular e hipotonía10.
Clínicamente observamos que los territorios afectados más frecuentes fueron los nervios de CPE y el cubital, y con menor frecuencia, los de mediano y el radial, con un solo caso de plexopatía braquial. Ello contrasta con lo descrito en la literatura, en la que por frecuencia los más afectados son el CPE, cubital y el plexo braquial4,12,13. Esta última afectación puede ser recurrente y habitualmente va asociada a otros síntomas. La presentación de plexopatía braquial como único síntoma de la enfermedad, como en nuestro caso, es inhabitual14. La literatura refiere que, de forma infrecuente, se pueden afectar otros nervios como el facial, trigémino, hipogloso y recurrente15. No hemos encontrado este tipo de afectación en nuestra serie.
En cuanto a la forma de presentación, además de los 2casos asintomáticos y los 13 con presentaciones típicas, 5 de nuestros pacientes empezaron de forma atípica. Esta proporción en torno al 25% es similar a la descrita en otras series4,13. Las diversas formas atípicas en la NHPP las clasificaron Pareyson en 199616 y Mouton en 1999,13 en 5grupos: 1) forma sensitiva recurrente posicional, 2) forma progresiva con alteración del CPE, 3) PNP S-M crónica, 4) PNP S-M tipo CMT y 5) la polirradiculoneuropatía desmielinizante crónica. Pou Serradell en 20024 añadió el síndrome de compresión crónica troncular (pluricanal), síndrome de debilidad crónica monotroncular no evolutiva y atrofia distal monomiélica. También se han descrito casos de afectación del sistema nervioso central como la disminución del volumen de la sustancia blanca con deterioro cognitivo15, neuropatía sensitivo motora hereditaria tipo viii (PNP más síndrome cerebelo-extrapiramidal)17 así como una niña de 7 años con alteración de la marcha, pies cavos, escoliosis y tortícolis, sin episodios de parálisis18. Las presentaciones atípicas en nuestra serie fueron síntomas sensitivos recurrentes en 3 casos, PNP S-M crónica en uno y mononeuropatía no evolutiva en otro. Queremos destacar la asociación familiar de diferentes presentaciones en miembros de una misma familia, en la que uno presentaba síntomas sensitivos recurrentes, otro mononeuropatía no evolutiva y el tercero, en cambio, una forma típica de la NHPP. Esta variabilidad fenotípica intrafamiliar no es común observarla19-21.
Independientemente de la presentación clínica, e incluyendo a los asintomáticos, todos los pacientes a los que se les realizó el estudio neurofisiológico mostraron alteraciones de la conducción nerviosa. Los pacientes de nuestra serie muestran porcentajes de afectación de nervios sensitivos con un rango de 84-94%, mayores que el rango de afectación motora (33-97%). Diferentes series muestran cierto predominio de afectación motora4,12,16,22,23,29 o, más infrecuentemente, predominio de afectación sensitiva22,24,25. Esta variabilidad puede deberse a la escasa muestra utilizada24,25 o al escaso número de nervios sensitivos evaluados2,8,13. Nuestro protocolo de exploración, además de los nervios sensitivos habitualmente explorados (mediano, cubital y sural), incluye la evaluación del peroneo superficial, hecho que puede explicar el mayor porcentaje de afectación sensitiva en nuestra serie.
Respecto a la alteración de la conducción nerviosa motora, fue principalmente alteración en lugares susceptibles de compresión. Se encontraron como parámetros de afectación más frecuentes la VCM del cubital sobre codo y la LDM del mediano y CPE. Dichos hallazgos concuerdan con los de la mayoría de las publicaciones2,3,8,11-13,16,23. Se ha descrito el aumento desproporcionado de la LDM respecto a la VCM como hallazgo característico del estudio de la conducción sensitiva motora en la NHPP25. Esta afirmación resulta controvertida30, dado que este hecho no puede extrapolarse a todos los nervios motores. Ocurre en determinados nervios como el mediano y el CPE, que son susceptibles de afectarse distalmente por movimientos repetitivos30. Nuestra serie confirma estos hallazgos en los nervios mediano y CPE, no observados en el cubital y tibial posterior.
Otra alteración de la conducción motora descrita como parámetro en el diagnóstico de la NHPP es la presencia de bloqueos de la conducción en los sitios de atrapamiento. En nuestros pacientes encontramos porcentajes más altos de lo habitual13. Destacamos un caso de bloqueo subclínico de la conducción del CPE izquierdo, que evolutivamente fue persistente, similar a otros casos descritos31. El mecanismo que conduce al bloqueo de la conducción en la NHPP no se conoce exactamente. Estudios de la excitabilidad axonal han demostrado alteraciones en el umbral del estímulo eléctrico, más pronunciados en la muñeca y también detectables en el codo. Los autores indican que las anomalías estructurales en los nodos de Ranvier pueden predisponer al bloqueo de la conducción en respuesta a la presión o estiramiento32.
Aunque en el diagnóstico de la NHPP el análisis del ADN es una herramienta objetiva y con elevada rentabilidad diagnóstica, proponemos resaltar que, dado el carácter hereditario de la neuropatía, el estudio neurofisiológico, además de su utilidad en pacientes con diagnóstico confirmado, es capaz de detectar a pacientes asintomáticos, por lo que debería realizarse búsqueda activa de sujetos de riesgo2 en familiares de pacientes afectos de NHPP11.
ConclusiónBasados en nuestros hallazgos, la alteración generalizada de la VCS, la disminución de la VCM del cubital sobre codo y el aumento de la LDM del mediano y CPE son las principales características neurofisiológicas en la NHPP con mayor afectación sensitiva en nuestra serie, lo que permite detectar a pacientes con clínica típica de NHPP, así como a pacientes con presentaciones atípicas e incluso a portadores asintomáticos. Dada la variedad fenotípica de esta entidad, esto convierte al estudio neurofisiológico en una herramienta tremendamente útil en el diagnóstico de NHPP.
Conflicto de interesesConfirmamos que no existe ningún conflicto de intereses que divulgar.

