






Los linfomas primarios del sistema nervioso central son una variedad poco frecuente de linfomas no hodgkinianos que constituyen alrededor del 4% de los tumores del sistema nervioso central.
Pacientes y métodosrealizamos una revisión retrospectiva de 24 pacientes diagnosticados de linfoma primario del sistema nervioso central entre enero de 1990 y diciembre de 2010. Todos los pacientes fueron diagnosticados con resonancia magnética y confirmados quirúrgicamente.
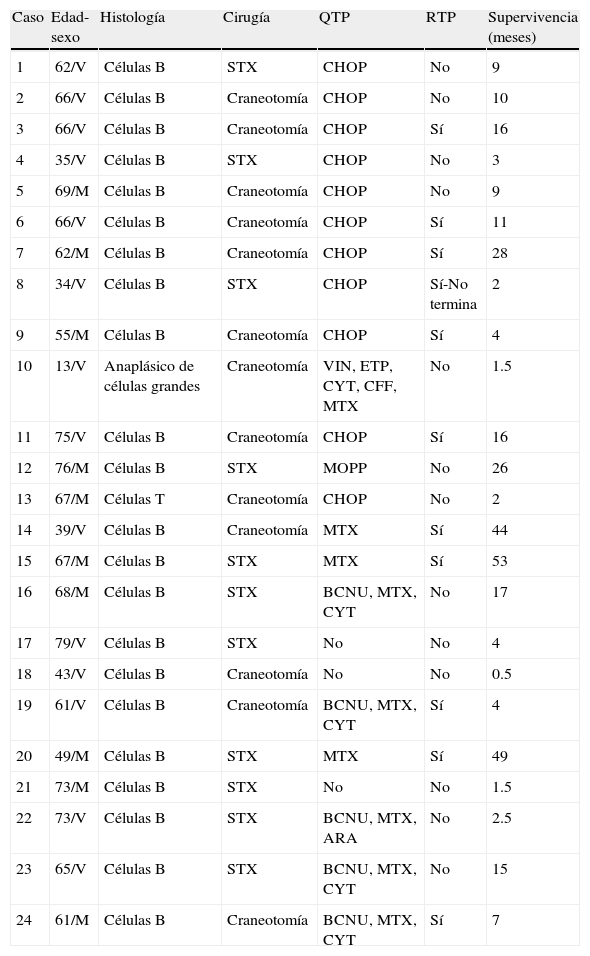
ResultadosDe los 24 pacientes analizados, 4 presentaban inmunodeficiencia. La media de edad era de 59,3 años (intervalo 13-79) y la relación entre varones y mujeres de 1 a 1,1. El deterioro cognitivo (33,4% de los pacientes) y la cefalea (22,5%) fueron los signos de presentación más frecuentes. El diagnóstico se realizó en 13 casos (54%) tras llevar a cabo una craneotomía y en los otros 11 (46%) mediante biopsia estereotáctica. La distribución histológica mostró que 22 casos (91,6%) eran linfomas tipo B, un caso un linfoma anaplásico de células gigantes y el otro correspondió a un linfoma de células T. La supervivencia media fue de 12,8 meses y a un año del 37,5%.
ConclusionesLos linfomas cerebrales primarios se presentan alrededor de la sexta década de la vida y clínicamente se manifiestan con deterioro cognitivo, cefalea y déficits neurológicos focales. El 75% de los pacientes (18 casos) presentaban únicamente una lesión intracraneal y el restante 25% (6 pacientes) entre 2 y 4 lesiones. El estado clínico preoperatorio constituye el factor pronóstico más importante.
Primary central nervous system lymphoma is a rare subtype of extranodal non-Hodgkin lymphoma that accounts for 4% of central nervous system tumours.
Patients and methodsRetrospective review of 24 patients diagnosed with primary central nervous system lymphoma between 1990 and 2010. All patients were diagnosed using magnetic resonance imaging and the diagnosis was confirmed surgically.
ResultsOf the 24 patients analysed, all except 4 were immunocompetent. Median age at diagnosis was 59.3 years (range 13-79) and the sex ratio (male to female) was 1:1.1. Cognitive decline (in 33.4%) and headache (in 25%) were the most common complaints. Diagnosis was performed In 13 cases (54%) following craniotomy and in the other 11 cases (46%) after stereotactic biopsy. Breakdown by pathology was as follows: 22 cases of B-cell lymphoma (91.6%), 1 case of anaplastic large-cell lymphoma, and 1 case of T-cell lymphoma. Mean survival time was 12.8 months with an overall 1-year survival rate of 37.5%.
ConclusionsPrimary central nervous system lymphoma often presents in the sixth decade with cognitive decline, headache, and focal neurological deficits. A single intracranial lesion was present in 75% of the patients (18 cases), and the remaining 25% (6 cases) had between 2 and 4 lesions. Preoperative clinical status was the most important factor determining prognosis.
El linfoma primario del sistema nervioso central (LPSNC), es un linfoma no-hodgkiniano que se origina en el cerebro, ojos, leptomeninges o médula espinal sin evidencia de linfoma sistémico en el momento del diagnóstico. Se trata habitualmente de tumores originados en células de tipo B y que resultan difícilmente diferenciados microscópica e inmuhohistoquímicamente de los linfomas sistémicos no hodgkinianos. En contraposición, los linfomas cerebrales secundarios se producen por una extensión o diseminación en el sistema nervioso central (SNC) de un linfoma sistémico1.
Los LPSNC constituyen alrededor del 4% de todos los tumores cerebrales primarios y entre el 1-2% de todos los linfomas. Su incidencia ha crecido lentamente en las últimas décadas debido a una mayor esperanza de vida de la población general y a la existencia cada vez mayor de pacientes inmunodeprimidos2.
La primera descripción fue realizada por Bailey3 en 1929, denominándolos «sarcomas perivasculares» debido a que las células tumorales tienden a rodear los vasos sanguíneos. En 1938, Yuile4 los denominó «sarcomas de células reticulares», y en 1948, Russell y Rubinstein5 introducen el término «microgliomas». La denominación actual se debe a Henry et al.6, quienes en 1974 los diferencian de los linfomas sistémicos y los denominan linfomas primarios del SNC. Posteriormente Rappaport7, en su clasificación de los linfomas, los incluye dentro del grupo de los linfomas no hodgkinianos.
Presentamos una revisión de 24 pacientes diagnosticados de LPSNC tratados en los últimos 21 años, y con un seguimiento mínimo de 12 meses, analizando sus características clínicas, neurorradiológicas, los aspectos relacionados con el tratamiento y su evolución.
Pacientes y métodosRealizamos un estudio retrospectivo descriptivo sobre los pacientes diagnosticados en nuestro servicio de neurocirugía, de linfoma primario cerebral entre los años 1990 y 2010.
Analizamos las características demográficas y clínicas, las técnicas de diagnóstico neurorradiologico empleadas, el tratamiento quirúrgico y oncológico practicado, así como su evolución. Para la valoración clínica de los pacientes se empleó la escala de Karfnosky (KPS). Además, en todos los pacientes, se realizó un estudio sistémico mediante análisis de médula ósea, ecografía abdominal y tomografía computarizada (TC) toracoabdominal como parte del estadio del tumor.
La confirmación del tumor se llevó a cabo en todos los casos mediante biopsia estereotáctica o por craneotomía. Se pudo realizar el seguimiento de todos los pacientes al menos durante 12 meses.
ResultadosLa serie consta de 24 pacientes (13 varones y 11 mujeres) con una media de edad de 54,7 años (intervalo 13-79). En los pacientes con sida, la edad media fue de 37,7 años, ascendiendo a 60 años en el resto de los pacientes (tabla 1).
Características histológicas, tratamientos recibidos y supervivencia de los pacientes con LPSNC
| Caso | Edad-sexo | Histología | Cirugía | QTP | RTP | Supervivencia (meses) |
| 1 | 62/V | Células B | STX | CHOP | No | 9 |
| 2 | 66/V | Células B | Craneotomía | CHOP | No | 10 |
| 3 | 66/V | Células B | Craneotomía | CHOP | Sí | 16 |
| 4 | 35/V | Células B | STX | CHOP | No | 3 |
| 5 | 69/M | Células B | Craneotomía | CHOP | No | 9 |
| 6 | 66/V | Células B | Craneotomía | CHOP | Sí | 11 |
| 7 | 62/M | Células B | Craneotomía | CHOP | Sí | 28 |
| 8 | 34/V | Células B | STX | CHOP | Sí-No termina | 2 |
| 9 | 55/M | Células B | Craneotomía | CHOP | Sí | 4 |
| 10 | 13/V | Anaplásico de células grandes | Craneotomía | VIN, ETP, CYT, CFF, MTX | No | 1.5 |
| 11 | 75/V | Células B | Craneotomía | CHOP | Sí | 16 |
| 12 | 76/M | Células B | STX | MOPP | No | 26 |
| 13 | 67/M | Células T | Craneotomía | CHOP | No | 2 |
| 14 | 39/V | Células B | Craneotomía | MTX | Sí | 44 |
| 15 | 67/M | Células B | STX | MTX | Sí | 53 |
| 16 | 68/M | Células B | STX | BCNU, MTX, CYT | No | 17 |
| 17 | 79/V | Células B | STX | No | No | 4 |
| 18 | 43/V | Células B | Craneotomía | No | No | 0.5 |
| 19 | 61/V | Células B | Craneotomía | BCNU, MTX, CYT | Sí | 4 |
| 20 | 49/M | Células B | STX | MTX | Sí | 49 |
| 21 | 73/M | Células B | STX | No | No | 1.5 |
| 22 | 73/V | Células B | STX | BCNU, MTX, ARA | No | 2.5 |
| 23 | 65/V | Células B | STX | BCNU, MTX, CYT | No | 15 |
| 24 | 61/M | Células B | Craneotomía | BCNU, MTX, CYT | Sí | 7 |
ARA: arabinosido; BCNU: carmustina; CFF: ciclofosfamida; CHOP: ciclofosfamida+doxorrubicina+procarbacina+prednisona; CYT: citarabina; ETP: etoposido; LPSNC: linfoma primario del sistema nervioso central; MOPP: clormetina+vincristina+procarbacina+prednisona; MTX: metotrexate; QTP: quimioterapia; RTP: radioterapia; STX: biopsia estereotáctica; VIN: vincristina.
La forma más frecuente de presentación fue el deterioro cognitivo en 8 pacientes (33,4%), seguida de la cefalea con 6 casos (25%), el déficit motor en 5 (20,8%), y las crisis comiciales en los restantes 5 pacientes (20,8%). Cuatro pacientes sufrían sida.
De acuerdo con la KPS, 14 pacientes se encontraban entre 90-100 en el momento del diagnóstico, 6 tenían entre 80-70, y 4 pacientes se encontraban entre 60-50.
Técnicas de diagnósticoEn todos los casos se realizó TC como técnica de diagnóstico inicial y resonancia magnética (RM) como método complementario. En la TC, 12 casos (50%) tenían características de hipodensidad, 8 (33,3%) eran hiperdensos y 4 (16,7%) isodensos. En 16 casos la captación de contraste era intensa, de ellos 3 de modo periférico (captación en anillo), y en 8 casos la captación era moderada.
En RM, en secuencias T1, 13 casos (54%) se mostraban como hipointensidad, 8 (33,3%) como isointensidad y 3 (12,7%) hiperintensidad moderada en relación con la sustancia gris; en secuencias T2, 21 casos (87,5%) se presentaban como hiperintensidad y en los 3 restantes (12,5%) la señal era heterogénea. En todos los casos la captación de gadolinio era entre moderada e intensa (fig. 1).
Localización, D. infiltración del cuerpo calloso..")
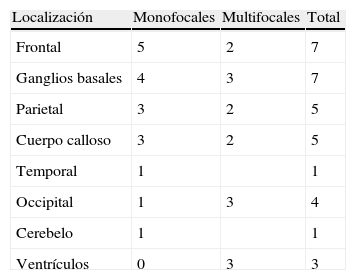
En total hemos identificado 33 lesiones intracraneales; 18 pacientes (75%) presentaban lesión única y 6 (25%) entre 2-4 lesiones (tabla 2). El hemisferio más afectado fue el derecho, donde se localizaban 16 tumores (48,6%), en el izquierdo se identificaron 12 (36,3%) y las 5 restantes (15,1%) estaban ubicadas en: cuerpo calloso (3 casos), hemisferio cerebeloso (un caso) y ventrículos laterales (una lesión) (fig. 2). Los 4 pacientes con sida presentaban lesión única (2 lobares, una hemisferio cerebelosa y una en ganglios basales).
.")
Tras el diagnóstico histológico, en 18 pacientes se realizó punción lumbar para analítica bioquímica y citológica del líquido cefalorraquídeo (LCR) (los restantes 6 pacientes fueron tratados oncológicamente en otros hospitales). En 5 casos se encontraron alteraciones en la proteinorraquia con cifras que oscilaron entre 0,6-1,45 g/l, en 3 de estos casos existía celularidad aumentada (12-44 células). En 5 de 19 casos (26%) se demostró la presencia de células malignas.
TratamientoEn 13 pacientes (54%) se realizó una craneotomía con exéresis amplia de la lesión (total en 12 casos) y en los otros 11 casos (46%) el diagnóstico definitivo se obtuvo mediante una biopsia esterotáctica (fig. 3).
Histología.")
El estudio anatomopatológico evidenció que 22 linfomas eran subtipo B, uno de células T y uno correspondió a linfoma anaplásico de células grandes (tabla 1).
Tratamiento oncológicoSe realizó algún tratamiento oncológico en 21 pacientes (3 pacientes rechazaron cualquier tipo de tratamiento complementario). El tratamiento recibido fue muy variado (los pacientes fueron tratados en diferentes servicios de hematología u oncología de la comunidad autónoma) y se ha modificado a lo largo de los años. En los pacientes más antiguos de la serie se administró el protocolo terapéutico consistente en CHOP (ciclofosfamida, doxorrubicina, vincristina y prednisona) (11 casos) o MOPP (clormetina, vincristina, procarbacina y prednisona) (un caso); en los restantes 9 pacientes se emplearon diferentes pautas de quimioterapia que aparecen recogidas en la tabla 1 y en todas ellas estaba incluido el metotrexate (MTX).
Se aplicó radioterapia en 14 pacientes; en 5 casos no se propuso debido a la mala situación clínica y la poca expectativa de supervivencia (casos 4, 10, 13, 18 y 21) y los restantes 5 casos no aceptaron esta terapia. La dosis empleada osciló entre 42-50,5Gy y en todos los casos se realizó exclusivamente a nivel craneal.
EvoluciónEn el momento de la revisión de las historias clínicas (noviembre de 2011) habían fallecido 23 pacientes, y únicamente una paciente de 49 años seguía viva con un seguimiento de 49 meses. La supervivencia media fue de 12,8 meses (intervalo 0,5-53). En los 4 pacientes con sida, la supervivencia osciló entre 2 y 44 meses (media 12,2), siendo en el resto de los pacientes de 15,5 meses (intervalo 0,3-64). Al año de seguimiento seguían vivos 9 pacientes (37,5%).
En los pacientes menores de 55 años la supervivencia media fue de 16,6 meses, siendo en los mayores de 55 años de 13. Con relación al estado clínico, en los pacientes con un Karfnosky≥80 la supervivencia media fue de 21,2 meses, reduciéndose a 7,1 en el resto.
DiscusiónLos LPSNC representan alrededor del 4% de todos los tumores cerebrales primarios y el 1-2% de los linfomas malignos no hodgkinianos, sin embargo, hasta un 10% de los casos de linfoma de cualquier localización puede cursar con algún tipo de afectación neurológica8. En las últimas décadas se ha producido un crecimiento lentamente progresivo en su incidencia, llegando a 30 casos/106 habitantes/año1, y se han convertido en el segundo tumor cerebral maligno más frecuentes en EE. UU. solamente por detrás de los gliomas9. Este incremento se debe, por un lado, al creciente número de pacientes inmunodeprimidos entre los que se incluyen enfermos trasplantados, oncológicos, o aquellos con sida; el otro gran motivo que contribuye a su incremento es la prolongación de la expectativa de vida, ya que el LPSNC se ha triplicado entre los individuos sanos mayores de 60 años sin conocerse claramente el motivo1,9,10. Entre los pacientes con sida, se calcula que el riesgo de padecer un LPSNC a lo largo del curso de la enfermedad es del 2-6%; entre los pacientes que reciben inmunosupresión farmacológica tras ser sometidos a un trasplante alogénico de órgano; este riesgo es del 1-5%, mientras que los que sufren algún tipo de inmunodeficiencia congénita, tienen un riesgo del 4%11. En nuestra serie sorprendentemente no hemos tenido ningún caso de paciente inmunodeprimido farmacológicamente, sin que sepamos a ciencia cierta cuál puede ser la explicación, aunque podría tratarse de pacientes con mala expectativa de supervivencia y se excluyera la indicación de biopsia cerebral.
No se encuentran diferencias significativas en la incidencia por sexos, si bien en la mayoría de las series, como en la nuestra, existe un ligero predominio de varones, debido posiblemente a una mayor incidencia de sida en estos. El pico de incidencia se sitúa en la sexta década de la vida, existiendo otro pico en la tercera-cuarta, correspondiendo a pacientes con algún tipo de inmunodeficiencia1.
La forma de presentación no se diferencia de otros procesos expansivos intracraneales, aunque en los linfomas son más frecuentes la presencia de deterioro cognitivo y cefalea, aunque pueden presentar otros síntomas como déficits neurológicos o crisis. Hasta un 10-15% de pacientes pueden presentarse con afectación ocular (uveítis o linfoma de vítreo), y en nuestra serie únicamente una paciente joven presentó a los 18 meses de evolución de la enfermedad un cuadro de infiltración vítrea.
En la TC, los LPSNC se muestran como tumoraciones únicas o múltiples, redondeadas u ovaladas, bien delimitadas y habitualmente de carácter hiperdenso12. Se encuentran rodeadas de una hipodensidad, correspondiente a edema13. Tras la administración de contraste, se observa de moderado a marcado realce. Los tumores de localización central tienen, con mayor frecuencia, una captación homogénea y los de localización periférica un realce en anillo, siendo esta forma de captación la más frecuente en los tumores de localización infratentorial14. Aunque en nuestros 4 casos con sida las lesiones eran únicas, los pacientes inmunodeprimidos presentan con mayor frecuencia localizaciones múltiples (hasta el 50% de los casos) y, en estos, las lesiones tienen un mayor carácter infiltrativo15. En el 75% de los casos se encuentra infiltración meníngea16 y es importante en estos casos diferenciarlos de los tumores primarios de meninges o de los linfomas de cráneo que posteriormente infiltran las meninges17.
En la RM se manifiestan como lesiones hipointensas en secuencias T1 e isointensas en T2, respecto a la sustancia gris, aunque no es raro que adopten otros tipos de señal18. Con la administración de gadolinio se realzan de forma importante, delimitando el contraste los márgenes del tumor, y separa el núcleo sólido del tejido edematoso adyacente que no se realza15.
En los estudios de difusión de imagen presentan áreas de restricción en el 90% de los casos en los estudios preoperatorios y aunque esta restricción también puede observarse en otros procesos expansivos como gliomas o metástasis, en los casos de LPSNC es de una manera muy acusada y el coeficiente de difusión aparente es menor19. En un reciente estudio de Barajas et al.20, se indica que el coeficiente de difusión aparente antes del tratamiento podría tener un valor predictivo en este tipo de linfomas. En la espectroscopia por RM se identifica una disminución de la concentración de N-acetilaspartato y una elevación de los niveles de lípidos, colina y el índice colina/creatinina20,21. Los estudios con tomografía por emisión de positrones (PET) tienen una sensibilidad diagnóstica del 100% independientemente del método empleado22 y el hallazgo característico es la presencia de lesiones hipermetabólicas20. Este hipermetabolismo es importante para hacer el diagnóstico diferencial en pacientes inmunocomprometidos en los que se puede sospechar la existencia de un proceso infeccioso o parasitario, en los cuales se observa un proceso hipometabólico20.
Macroscópicamente son tumores parecidos a los gliomas de alto grado y presentan, como ellos, zonas infiltrativas y áreas de necrosis1. Pueden asentar tanto en la sustancia gris como en la blanca, y en más del 80% de los casos se sitúan en la región supratentorial, en la sustancia gris profunda y con tendencia a localizarse en las inmediaciones del sistema ventricular. Microscópicamente están formados por masas de células linfoides, con gran densidad celular en las partes centrales del tumor, donde llega a desaparecer la estructura del parénquima cerebral. Las células se disponen alrededor de los vasos, ocupando y ensanchando los espacios de Virchow-Robin, donde separan las fibras de reticulina e inducen la formación de nuevas fibras23. Aunque se trata de masas bien delimitadas, no es raro encontrar invasión tumoral más allá de los márgenes macroscópicos del tumor. Según Isaacson y Norton24 el 75-90% son linfomas difusos de célula grande B (centroblásticos y, sobre todo, inmunoblásticos) (92% en nuestra serie), un 5% linfomas tipo Burkitt y un 10-25% son linfomas de bajo grado, sobre todo linfomas linfoplasmocitoides y en menor frecuencia linfomas centrofoliculares difusos.
Patogénicamente, se describen 4 patrones:
- I.
Nódulos solitarios (56%) o múltiples de localización intracraneal (26%), que es la forma más frecuente6,15,25.
- II.
Afectación meníngea difusa o lesiones periventriculares con infiltración del espacio subaracnoideo en el 20-50% de los casos26 o ependimaria27.
- III.
Depósitos vítreos o uveales (15%)28,29.
- IV.
Masas espinales de localización intradural30–32. Se trata de una forma extremadamente rara y representa menos del 1% de todos los LPSNC.
Desde el punto de vista inmunofenotípico, la mayoría expresan antígenos pan B (CD20 y CD79a), con expresión monoclonal de inmunoglobulina de superficie, sobre todo IgMk. Los LPSNC asociados a inmunosupresión suelen expresar proteínas de latencia de membrana o antígenos nucleares del virus de Epstein-Barr, a diferencia de lo que ocurre en pacientes no inmunodeprimidos. En los linfomas de células T encontramos positividad para CD45RO, y CD38. En algunos estudios la sobreexpresión Bcl-6 es variable y tiende a asociarse con mejor pronóstico. Por su parte, la expresión P57 y C-myc comporta peor pronóstico33.
En los estudios de genética molecular los linfomas expresan reagrupamientos clonales del gen de las inmunoglobulinas (en el caso de los linfomas B) o del gen del receptor T (en los casos de fenotipo T). Se han descrito pérdidas de material genético que se localizan sobre todo en el cromosoma 6 y se han relacionado con un peor pronóstico y las ganancias más habituales son a nivel 12q. La metilación del promotor del gen del transportador del folato reducido se ha relacionado con resistencia al MTX34.
Debido a que en el sistema nervioso no existe tejido linfoide, la patogenia de estos linfomas es objeto de controversia continuada. Hay diversas hipótesis, todas ellas especulativas, ya que existen pocos estudios con hipótesis sólidas35. Las células linfomatosas se originarían en cualquier punto del organismo (fuera del SNC), pero se desarrollarían en el cerebro tras haber recibido receptores «homing» específicos para los endotelios cerebrales, y una vez situados a este nivel, no podrían ser destruidos por el sistema inmunológico. Existe otra teoría según la cual existiría una lesión inflamatoria previa que condicionaría una reacción policlonal de células linfoides en el seno de la cual podría surgir un clon neoplásico, tal y como se supone ocurre en otros linfomas11. Un tercer mecanismo apuntado incide en que las células linfomatosas generadas fuera del SNC se erradican sistemáticamente con un sistema inmunitario competente, pero proliferan en el cerebro15.
El tratamiento se inicia después del procedimiento quirúrgico que lleva al diagnóstico. En nuestra experiencia se realizó craneotomía en 13 casos (54.1%) y biopsia estereotáctica en 11, sin embargo en la mayoría de las series el diagnóstico suele realizarse mediante biopsia debido a que muchos de ellos tienen una localización profunda o multicentrica; además no se ha podido demostrar que una resección amplia tenga mejor pronóstico que la realización de una biopsia. Es importante considerar que esta técnica es menos invasiva, tiene un rendimiento diagnóstico superior al 95% y el índice de complicaciones es menor que el de una craneotomía36,37. La resección por craneotomía puede ocasionar déficits neurológicos, retrasa el inicio del tratamiento oncológico y no mejora la supervivencia38,39, y únicamente estaría indicada en caso de lesiones únicas, fácilmente accesibles, con la seguridad de no aumentar la morbilidad. Cuando se realiza una biopsia estereotáctica es importante reducir la terapia esteroidea, ya que, debido a su efecto citolítico, puede producir una reducción en el tamaño del tumor llegando en ocasiones a desaparecer por completo (tumor fantasma)40. Aunque algunos autores indican que se puede hacer un buen diagnóstico sin suprimir los esteroides41, existe un consenso generalizado en que estos se deben interrumpir entre 5 y 10 días antes de realizar la biopsia. En nuestra experiencia siempre realizamos la toma de muestra después de al menos 10 días sin esteroides, lo que permitió tener un diagnóstico definitivo en la primera biopsia en todos los casos.
Tras el diagnóstico del tumor es imprescindible la realización del estudio de extensión; la afectación de diferentes áreas del SNC como ojos, meninges o nervios craneales no implica un estadio más avanzado ni un peor pronóstico38. Se calcula que entre el 4-12% de los linfomas considerados inicialmente primarios del SNC van a tener afectación sistémica42. La citología del LCR aparece como prueba de extensión en todos los protocolos, sin embargo en nuestra serie solo fue positiva en el 27,7% de los pacientes; la afectación meníngea incrementa los índices de positividad en los resultados43. La analítica del LCR es importante no solo para orientar el diagnóstico sino también porque tiene connotaciones terapéuticas. La presencia intratecal de células tumorales justifica el empleo de terapia intratecal8. Si no se ha realizado previamente, es fundamental la RM de todo el neuroeje. El rastreo con PET podría sustituir a la práctica del TC torácica y abdominal y la biopsia de medula ósea, al demostrase más sensible para detectar pequeños focos de linfoma sistémico22,44. Aunque se han descrito algunos casos de remisión espontanea de un LPSNC sin terapia esteroidea previa45, se trata de una situación excepcional que, en nuestra opinión, no debe retrasar el inicio de la terapéutica. El tratamiento oncológico se basa en la corticoterapia, quimioterapia y radioterapia, ya que muestran una especial sensibilidad a estas terapias1.
Los corticoides son citolíticos para el LPSNC y pueden causar una respuesta parcial o total en el 40% de los pacientes no inmunodeprimidos; sin embargo esta, es de poca duración y no tiene efecto curativo ni predice una mejor evolución. La combinación de quimioterapia y radioterapia se considera actualmente el mejor sistema de tratamiento. Los esquemas tradicionales de linfoma sistémico (CHOP) no son efectivos al no pasar la barrera hematoencefálica (BHE)38. Aunque el tratamiento con CHOP produce una buena respuesta inicial al estar la mayoría del tumor no protegido por la BHE, los siguientes ciclos de CHOP no consiguen erradicar los restos del linfoma, debido probablemente a que con las primeras dosis se normaliza la BHE38. En nuestra experiencia, se han empleado muy diversas pautas de tratamiento motivado, en primer lugar, por el amplio período de tiempo analizado y, en segundo lugar, porque 6 de los pacientes provenían de otros hospitales de la Comunidad y solo acudieron a nuestro servicio para la realización de la craneotomía o de la biopsia, siguiendo posteriormente el tratamiento en su hospital de procedencia.
En los paciente no inmunodeprimidos el MTX y la citarabina son los fármacos más activos, y los regímenes de tratamiento que incluyen estos fármacos y radioterapia consiguen una tasa de respuesta cercana al 80% y una supervivencia media de 3 años14.
Actualmente existen ensayos de tratamiento con temozolamida (TMZ); esta tiene la ventaja de una buena tolerancia y se obtienen índices de remisión completa al año del 31%, en pacientes refractarios a MTX46. La combinación de TMZ con MTX a altas dosis está teniendo unos resultados alentadores incluso en pacientes ancianos47. La monoterapia con TMZ se asocia con un 47% de remisión competa al año y una supervivencia media de 21 meses en pacientes ancianos48.
El rituximab es un fármaco muy utilizado en linfomas periféricos, sin embargo, al tratarse de una macroproteína, no atraviesa bien la BHE. Se han publicado algunos estudios en monoterapia o combinados con MTX49. Aunque el nivel de evidencia sobre su empleo en LPSNC es bajo, su uso tiene cierto valor para algunos oncólogos aunque en general se piensa que solo debería emplearse en ensayos prospectivos38.
Algunos autores aconsejan el empleo de quimioterapia intraarterial, precedida del empleo de manitol para provocar una disrupción transitoria de la BHE y así poder aumentar la concentración de fármacos en el SNC50.
Aunque no hay estudios comparativos, los análisis retrospectivos de las series que han realizado tratamiento con MTX intratecal no demuestran una mejor supervivencia frente a series que no usaron este tratamiento51. En un reciente estudio retrospectivo de Sierra et al.52, en el que se analizan 69 pacientes con LPSNC en los cuales se empleó MTX intratecal en 39 casos y no se utilizó en los otros 30 pacientes, no encontraron diferencias estadísticamente significativas entre los 2 grupos.
La neurotoxicidad es un problema importante en los pacientes que reciben tratamiento combinado con quimio y radioterapia. La mayoría desarrollan un cuadro de inestabilidad, incontinencia de esfínteres y deterioro cognitivo, observándose en la RM datos de afectación difusa de la sustancia blanca y de atrofia cerebral. En pacientes mayores de 60 años el riesgo de neurotoxicidad es prácticamente del 100% y se desaconseja la radioterapia si el LPSNC está en remisión al acabar la quimioterapia. En un reciente estudio Correa et al.53 han observado que el deterioro cognitivo es más acusado en aquellos pacientes supervivientes tratados con radioterapia y MTX a altas dosis que en aquellos tratados únicamente con el MTX a dosis altas. Como alternativa, en un reciente estudio de Alimohamed et al.54, tratan 21 pacientes con LPSNC con altas dosis de thiotepa, busulfan, ciclofosfamida y trasplante autólogo de progenitores hematopoyéticos sin emplear radioterapia holocraneal. Observaron que ningún paciente desarrolló neurotoxicidad y que el 52% de los pacientes estaban vivos y libres de enfermedad a los 60 meses.
En el LPSNC la radioterapia constituye uno de los pilares del tratamiento. En estos pacientes se incluye la radiación craneal completa, ya que la radiación exclusivamente local se acompaña de mayores índices de recidiva. Aunque exista diseminación por el LCR no se aconseja la radiación espinal, ya que no aporta mayor supervivencia pero sí más morbilidad39 y esta solo estaría indicada en los casos raros de linfoma espinal1. La radioterapia como tratamiento único es pocas veces curativa, y cuando se ha empleado la supervivencia media osciló entre 10-18 meses55. Sin embargo sí puede estar indicada en casos de tratamiento paliativo o en pacientes con pequeños linfomas linfocíticos o linfoblásticos originados en las meninges38. La dosis de radioterapia es variable en diferentes estudios, pero suele oscilar entre 40-50Gy; el empleo de dosis mayores de 50Gy con el añadido de una sobreimpresión no ha demostrado una mayor eficacia y sí un incremento de la toxicidad38.
Otro aspecto controvertido es el tratamiento de las recaídas, aunque actualmente no hay una estrategia única, y el tratamiento debe considerarse en función de la edad del paciente, el estado funcional y los tratamientos previos. En paciente jóvenes con buen estado general se puede realizar un tratamiento de rescate que incluya autotrasplante. En un estudio de Soussain et al.56 de 43 pacientes con LPSNC recidivado, se trataron con 2 ciclos de citarabina y etopósido seguido de autotrasplante. Observaron que la supervivencia a los 2 años fue del 69% (76% para los que se trasplantaron en remisión completa).
En un reciente estudio multicéntrico de Thiel et al.57 en el que analizan a 551 pacientes con LPSNC de pacientes inmunocompetentes, fueron randomizados en 2 grupos, en el primer grupo fueron tratados con altas dosis de MTX y radioterapia y el otro grupo emplearon únicamente altas dosis de MTX. Observan que los pacientes tratados con radioterapia desarrollarían más toxicidad pero que entre ambos grupos no hay una diferencia significativa en cuanto a intervalo libre de enfermedad ni en la supervivencia
El pronóstico de los LPNC es mucho peor que el de los linfomas sistémicos58. La supervivencia media sin tratamiento es de 2-4 meses tras el diagnóstico59. Con radioterapia, la supervivencia es de 10 meses y con la adicción de quimioterapia y MTX intratecal se consiguen tasas de respuesta inicial del 85% y las recurrencias suelen aparecer entre los 15 y 45 meses del tratamiento1. En nuestra serie, los pacientes con sida tienen una supervivencia ligeramente inferior al resto de los pacientes y esta viene motivada en gran parte por el estadio evolutivo de la enfermedad basal1.
En cuanto al pronóstico, en nuestra experiencia el estado clínico preoperatorio constituye el factor más importante, llegando los pacientes con un Karfnosky≥80 a tener una supervivencia hasta 3 veces superior a aquellos con una puntuación inferior. Aunque en la literatura se indica que los pacientes con sida tienen una peor evolución, en nuestra experiencia la diferencia de supervivencia entre ambos grupos es poco importante (12,2 vs 15,3 meses) pero debe tomarse con cautela, ya que se trata de un grupo muy reducido de pacientes. En la literatura se han identificado 5 factores predictores independientes: edad, KPS, nivel de LDH en suero, proteinorraquia y afectación de estructuras profundas60. En función de estos factores, el Grupo de Estudio del Linfoma Extranodal distingue 3 grupos de riesgo basados en la presencia de 0-1, 2-3 o 4-5 de los factores61. Aunque no existe unanimidad entre los autores, también se consideran factores de buen pronóstico: la lesión intracraneal única, ausencia de tumor meníngeo o periventricular, ausencia de inmunodeficiencia, emplear tratamiento con quimio y radioterapia62,63.
Los LPSNC tienen 2 picos de incidencia alrededor de la cuarta y sexta décadas de la vida, correspondiendo los pacientes más jóvenes a aquellos con sida. La forma de presentación clínica más frecuente es el deterioro cognitivo, seguido de cefalea y déficits neurológicos focales. En nuestra experiencia el 75% de los pacientes presentaban una única lesión intracraneal. La mayoría de los LPSNC son histológicamente de células grandes tipo B y el estado clínico preoperatorio constituye el factor pronóstico más importante.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.

