



Prader-Willi syndrome is a neurodevelopmental genomic imprinting disorder with lack of expression of genes inherited from the paternal chromosome 15q11-q13 region usually from paternal 15q11-q13 deletions (about 60%) or maternal uniparental disomy 15 or both 15s from the mother (about 35%).1 The child usually presents with infantile hypotonia, poor sucking ability, failure to thrive, short stature, and hypogonadism/hypogenitalism. Associated remarkable features include hyperphagia, morbid childhood obesity, polyendocrinopathies (suggestive of hypothalamic dysfunction), and cognitive/behavioral disturbances.1,2
Hypothyroid myopathy is a typical clinical characteristic of patients with either primary or secondary, and even, subclinical hypothyroidism.3 On the other hand, Hoffman's syndrome, a rare form of hypothyroid myopathy, which is predominantly seen in adults with long-standing untreated primary hypothyroidism,4 and rarely in secondary hypothyroidism,5 is characterized by increased muscular mass (pseudohypertrophy), proximal muscle weakness, muscle stiffness, and cramps.3–5 Although the pathophysiology remains unclear, it is likely that deposition of glycosaminoglycans within muscle fiber cells increase the muscle size.3–5We report the case of a patient who presented with clinical, biochemical and electrophysiological features of Hoffman's syndrome following secondary thyroid insufficiency. Clinical history, physical examinations, and genetic studies further established the diagnosis of Prader-Willi syndrome.
A 20-year-old male from rural India came to our clinic due to voracious appetite, food craving, irresistible feeding, and excessive weight gain from two years of age, and absence of testes in scrotal sac. He could not suck properly during the neonatal period, but no document was suggestive of birth asphyxia. His four siblings died in utero (stillborn). Moreover one died after 10 days of birth who reportedly had facial dysmorphism. All his developmental milestones were delayed. He had been hospitalized several times for episodes of respiratory tract illness. There was also a history of passage of large volume, bulky stool 3–4 times a day. He started smoking daily at the age of four despite solid prohibition. He also had a history of excessive daytime somnolence, snoring, and mouth breathing. He also felt difficulties in getting up from the sitting/lying down position and getting upstairs, which was insidious in onset and slowly progressive over the last six years, and was associated with exercise intolerance, cramps, pain, and stiffness.
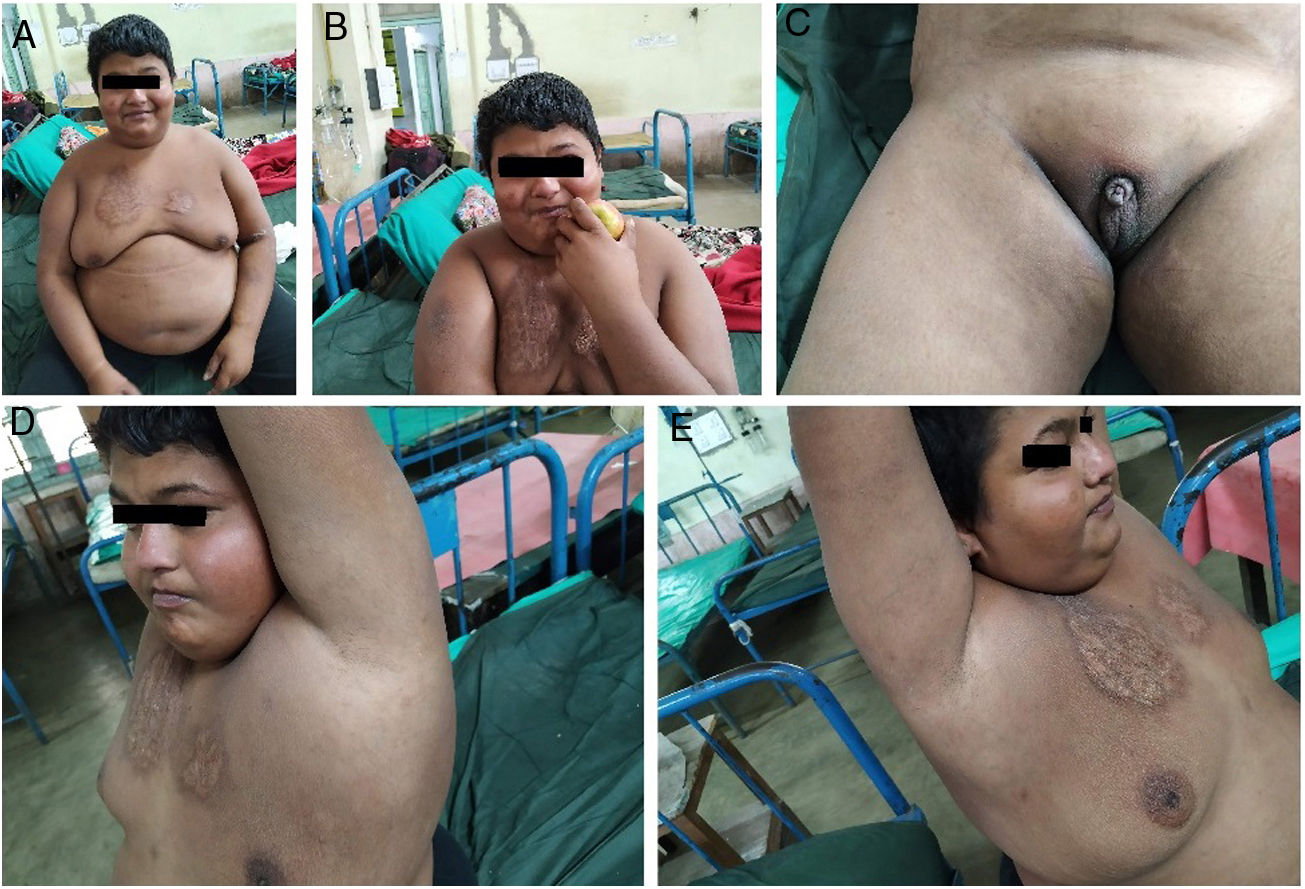
On general examination (Fig. 1), he was inattentive and sleepy all the time. He had a non-constitutional short stature (height 135cm), morbid obesity (body mass index=35.5), dry skin, high-pitched slurred voice, undescended testes, maldeveloped scrotum, lipomastia, and under-development of other secondary sexual characteristics. In addition, he had conjunctival congestion, facial plethora, poor-oral hygiene, acrodermatitis, and asymmetrical scar-like lesions over both sides of the anterior chest wall, which developed because of repeated episodes of “cigarette-burn” (Fig. 1). Neurological examination revealed flabby muscle bellies, significant muscle enlargement (pseudohypertrophy), proximal limb muscle weakness (affecting lower limbs more than upper limbs), myoedema, proximal-type gait, generalized hyporreflexia, and positive Woltman sign. Other vital signs were within normal limits. He had moderate deficits in intellectual capacities (intelligence quotient=40) and moderate deficits in the social adaptive functioning (social quotient=37).
, dry skin (A–E), small testes (C), maldeveloped scrotum (C), lipomastia (A), and under-development of other secondary sexual characteristics (C–E). Besides, he had conjunctival congestion, facial plethora (especially malar flushing) (B), asymmetrical scar-like lesions over both sides of the anterior chest wall (A–E), which developed due to repeated episodes of “cigarette-burn” (done mainly by addicted peer groups and sometimes self-inflicted) over a long period.")
The patient had morbid obesity (A), dry skin (A–E), small testes (C), maldeveloped scrotum (C), lipomastia (A), and under-development of other secondary sexual characteristics (C–E). Besides, he had conjunctival congestion, facial plethora (especially malar flushing) (B), asymmetrical scar-like lesions over both sides of the anterior chest wall (A–E), which developed due to repeated episodes of “cigarette-burn” (done mainly by addicted peer groups and sometimes self-inflicted) over a long period.
Complete blood cell count revealed isolated polycythemia with raised erythropoietin levels suggestive of acquired secondary polycythemia. Thyroid function tests revealed severe non-primary (central) hypothyroidism (thyroid-stimulating hormone=0.50mIU/mL, free T4<1.0pmol/L, and free T3<0.70pmol/L) with negative anti-thyroid antibodies. Other hormonal profiles revealed low levels of luteinizing hormone, follicle-stimulating hormone, testosterone, and insulin-like growth factor-1, and normal levels of prolactin and cortisol/adrenocorticotropic hormone. Serum creatine phosphokinase level was raised (4500U/L; normal<170). Needle electromyography from muscles of proximal upper and lower limbs showed low amplitude, short duration polyphasic motor unit action potentials, with early recruitment and without any spontaneous activity, suggestive of a myopathic pattern (lower limbs were affected more than upper limbs). The repetitive nerve stimulation test and computed tomographic scan of the thorax (to visualize thymus pathology) were normal. Anti-acetylcholine receptor, anti-muscle-specific kinase antibodies, and muscle-specific autoantibodies were negative. Clinical exome sequencing for muscular dystrophies, metabolic myopathies, and myasthenic disorders were negative. Histopathological confirmation with muscle biopsy with biochemical muscle enzyme assay was not performed due to lack of consent from both the patient and his guardian (mother) and deficit in infrastructural set up in our institution for performing such advanced muscle histopathological examinations. Abdominal ultrasound revealed small bilateral testes placed at respective inguinal canals near ipsilateral superficial inguinal rings; both scrotal sacs were empty. Pulmonary function tests revealed moderate obstructive airway disease. A sleep study was suggestive of obstructive sleep apnea. Echocardiography showed the presence of mild pulmonary arterial hypertension and mild pericardial effusion without any features of cardiomyopathy. Fluorescent in-situ hybridization analysis was performed on 20 metaphases, which documented Prader-Willi syndrome (Del 15q11-13) syndrome.
Thyroid hormone replacement therapy with levothyroxine was started, and gradually increased to 125 mcg/day over a period of three months. Although the neurodevelopmental disabilities persisted, hypothyroid myopathy improved (myoedema was non-elicitable; deep tendon reflexes normalized; motor strength increased; and subtle objective decrement in body mass index, as well as of the diameter of pseudo-hypertrophic muscle bellies were observed) after three months of therapy. In addition, serum CPK normalized and disappeared the pericardial effusion on echocardiography. His family refused to start growth hormone and testosterone replacement therapy as well as continuous positive airway pressure therapy for obstructive sleep apnea, despite counselling, quite understandably due to financial restraints.
Several endocrine disorders may be found in Prader-Willi syndrome, and thyroid hormonal imbalance is one of them.6 The prevalence of abnormal thyroid function in Prader-Willi syndrome patients varies from 2 to 32%.6 Primary hypothyroidism occurs when the thyroid gland does not respond to a thyroid-stimulating hormone released by the brain while central hypothyroidism occurs when there is a lack of thyroid-stimulating hormone production.7 In the PWS population, central hypothyroidism may be more prevalent due to brain dysfunction is known to be associated with this disorder.7 In our patient, short stature, morbid obesity, voracious eating, cryptorchidism, and hypogonadism maybe attributed to Prader-Willi syndrome, meanwhile hypothyroidism might have additionally contributed to non-constitutional short stature and morbid obesity, and have caused muscular weakness.
Motor problems in Prader-Willi syndrome start in infancy and persist throughout childhood and adulthood, contributing to lower scores in standardized motor performance tests and abnormal gait.8 These problems are presumed to be due to abnormal body composition.8 Indeed, in Prader-Willi syndrome there is an increase in fat mass and a decrease in lean body mass (probably related to hormonal deficiencies as a result of hypothalamic dysfunction and deficiency of growth, thyroid, gonadal and adrenal hormones), and possibly some degree of neuromuscular abnormality.8 Notwithstanding, although motor problems in Prader-Willi syndrome are quite typical, the exact pathogenetic mechanism behind the same remains unknown.8 Growth hormone deficiency and different structural and functional abnormalities of muscle (e.g., type-2 muscle fiber atrophy with type 2-B fiber deficiency increased immature type-2C muscle fibers, and decrease in CoQ10 levels in muscle tissue) may lead to decrease in muscle mass.8 However, only a decrease in muscle mass cannot explain the muscle weakness in Prader-Willi syndrome. Probably, hypotonia and muscle disuse during infancy may hamper motor cortex stimulation during the sensitive period thereby leading to motor problems later in life in Prader-Willi syndrome.
Although hypogonadism occurs in conjunction with proximal myopathy in proximal myotonic myopathy syndrome,9 negative family history, absence of myotonia, cataracts, and tremor excluded this possibility in our case. Other plausible differential diagnoses that were excluded with relevant clinical history/examinations and laboratory investigations are furnished briefly in Table 1.
Differential diagnoses of the case.
| Differential diagnoses | Points against this diagnosis |
|---|---|
| Limb-girdle muscular dystrophy (LGMD) | • The major features of LGMD are progressive wasting (atrophy) and weakness of the proximal muscles of the hip and shoulder areas. Our patient did not have muscle atrophy and shoulder girdle muscles had only subtle involvement.• Family history was negative.• No cardiac muscle involvement.• Improvement with levothyroxine replacement therapy.• Clinical exome sequencing for LGMD was negative. |
| Idiopathic inflammatory myopathies (IIMs) | • The IIMs are a heterogeneous group of diseases, collectively termed myositis, sharing symptoms of muscle weakness, fatigue, and inflammation. Other organs are frequently involved, supporting the notion that these are systemic inflammatory diseases. The IIMs can be subgrouped into dermatomyositis, polymyositis and inclusion body myositis.• The myositis-specific autoantibodies panel was negative.• No extra-muscular/systemic manifestations like that of IIMs were found in this patient.• No associated neoplastic process could be documented.• Improvement with levothyroxine replacement therapy.• Improvement without institution of immunomodulator therapy. |
| Myasthenic syndromes | • No ptosis, external ophthalmoplegia, bulbar weakness, and exertional fatiguability.• No diurnal variation in weakness.• Anti-acetylcholine receptor, anti-muscle specific kinase antibodies were negative.• Repetitive nerve stimulation test was negative.• CT scan of the thorax was negative for any thymus pathology.• Clinical exome sequencing for myasthenic disorders were negative.• Improvement with levothyroxine replacement therapy.• Improvement without institution of immunomodulator therapy. |
| Hypoandrogenism associated myopathy | • Hypogonadism was present in this patient since his birth, but the symptoms were relatively much later in onset; only to be manifested at early adulthood.• Improvement with levothyroxine replacement.• Improvement without replacement of testosterone. |
| Drug-induced myopathy | • No history of long-term intake of any drug that could cause myopathy. |
In our patient, the most plausible explanation is that Prader-Willi syndrome led to non-primary hypothyroidism, which went unnoticed for an extended period and in turn gave rise to Hoffman's syndrome, which responded remarkably with thyroid hormone replacement only. This latter syndrome is rare in itself and the underlying etiology (Prader-Willi syndrome) for this type of hypothyroid myopathy makes this case unique.
Authors’ contributionsAll authors contributed significantly to the creation of this manuscript; each fulfilled criteria as established by the ICMJE.
Data availability statementWritten informed consent was obtained from the patient participating in the study (consent for research).
Ethics statementInformed written consent was obtained from the patient involved in this study.
Study fundingNone.
DisclosuresR. Ghosh reports no disclosures relevant to the manuscript.
A. Ray reports no disclosures relevant to the manuscript.
D. Roy reports no disclosures relevant to the manuscript.
J. Benito-León reports no disclosures relevant to the manuscript.
Conflict of interestThe authors declare that they have no conflict of interest.
J. Benito-León is supported by the National Institutes of Health, Bethesda, MD, USA (NINDS #R01 NS39422), European Commission (grant ICT-2011-287739, NeuroTREMOR), the Ministry of Economy and Competitiveness (grant RTC-2015-3967-1, NetMD—platform for the tracking of movement disorder), and the Spanish Health Research Agency (grant FIS PI12/01602 and grant FIS PI16/00451).

