



Los pacientes con síndrome de Down (SD) presentan una demencia tipo Alzheimer (EA) asociada a la edad. Ambas patologías, con una base neuropatológica común, han sido asociadas a la epilepsia mioclónica de inicio tardío (LOMEDS). Esta entidad presenta alteraciones electroencefalográficas características en forma de descargas generalizadas de polipunta-onda.
MétodoPresentamos una serie de 11 pacientes con el diagnóstico de SD o EA que desarrollaron crisis epilépticas mioclónicas o tónico-clónicas generalizadas. En todos ellos, se realizó un seguimiento clínico y estudios de neuroimagen y poligrafía EEG.
ResultadosEn todos los casos, el deterioro cognitivo avanzó rápidamente tras el comienzo de la epilepsia, produciendo un incremento en el grado de dependencia. El hallazgo más común en el EEG fue un enlentecimiento de la actividad cerebral con ritmos theta y delta; además, en 8 pacientes se objetivaron descargas intercríticas generalizadas de polipunta-onda. En los estudios de neuroimagen se encontró atrofia cerebral cortical. El fármaco más eficaz en esta serie fue el levetiracetam.
ConclusionesLa asociación de epilepsia generalizada al SD de edad avanzada supone un epifenómeno en la evolución que marca un agravamiento rápidamente progresivo de las funciones cognitivas y motoras. Presenta unas características electroclínicas bien definidas y se comporta como una epilepsia mioclónica progresiva, que probablemente se relaciona con los cambios estructurales que caracterizan el parecido evolutivo del SD con la enfermedad de Alzheimer. El reconocimiento de este síndrome es importante, dado que tiene repercusiones pronósticas y requiere un tratamiento adecuado.
Patients with Down syndrome (DS) who exhibit Alzheimer disease (AD) are associated with age. Both diseases with a common neuropathological basis have been associated with late-onset myoclonic epilepsy (LOMEDS). This entity presents electroencephalogram features as generalized polyspike-wave discharges.
MethodWe present a series of 11 patients with the diagnosis of DS or AD who developed myoclonic seizures or generalized tonic-clonic seizures. In all cases, clinical and neuroimaging studies and polygraph EEG monitoring was performed.
ResultsIn all cases, cognitive impairment progressed quickly after the onset of epilepsy causing an increase in the degree of dependence. The most common finding in the EEG was a slowing of brain activity with theta and delta rhythms, plus intercritical generalized polyspike-waves were objectified in eight patients. In neuroimaging studies was found cerebral cortical atrophy. The most effective drug in this series was the levetiracetam.
ConclusionsThe association of generalized epilepsy with elderly DS represents an epiphenomenon in evolution which is associated with a progressive deterioration of cognitive and motor functions. This epilepsy has some electroclinical characteristics and behaves as progressive myoclonic epilepsy, which is probably related to the structural changes that characterize the evolutionary similarity of DS with AD. Recognition of this syndrome is important, since it has prognostic implications and requires proper treatment
La prevalencia de crisis epilépticas en pacientes con enfermedad de Alzheimer (EA) es más elevada que en la población normal1. La prevalencia de epilepsia en el síndrome de Down (SD) aumenta con la edad, alcanzando un 46% en pacientes de más de 50 años2. Del mismo modo que en la población sana, la incidencia de epilepsia focal de causa estructural en pacientes con SD se incrementa con la edad.
En el cerebro de pacientes con SD que desarrollan demencia o deterioro cognitivo leve se ha descrito la presencia de cambios anatomopatológicos compatibles con EA3. En su patogénesis desempeña un papel fundamental la trisomía 21, donde se encuentran los genes del péptido precursor del amiloide (PPA) —que lleva a una mayor producción de proteína amiloide— y el gen ß-site amyloid precursor protein cleaving enzyme 2 (BACE 2) —que produce péptidos amiloidogénicos constituyentes de las placas seniles. Otros factores, como los estrógenos, los alelos ApoE o los polimorfismos del gen de la proteína priónica (PRNP) están también implicados.
Se pensaba que la epilepsia era un fenómeno habitualmente tardío en la evolución de la EA. Sin embargo, Lozsadi y Larner4 describen que hasta el 6,8% de los pacientes con un posible diagnóstico clínico de EA presentan epilepsia, precisando de medicación antiepiléptica —en un 3,4% coincide el inicio de las crisis epilépticas con el inicio del deterioro cognitivo.
Se ha caracterizado un síndrome de epilepsia mioclónica en los pacientes con SD en el contexto de demencia tipo EA, con un carácter progresivo y coincidiendo con una aceleración importante en el deterioro cognitivo y en el grado de dependencia. Esta entidad fue descrita por primera vez en 1994 por Genton y Paglia5 y se caracteriza por mioclonías de predominio matutino, afectando especialmente a las extremidades superiores y que se asocian a descargas generalizadas de polipunta-onda en el EEG.
El presente estudio tiene por objetivo describir una serie de pacientes con SD o EA con mioclonías y crisis epilépticas tónico-clónicas generalizadas; con la finalidad de comprender mejor este cuadro y realizar un manejo adecuado de estos pacientes.
MétodoEstudio descriptivo basado en una revisión retrospectiva de una serie pacientes procedentes de 3 centros hospitalarios de Asturias (Hospital Alvarez Buylla, Hospital Valle del Nalón y Hospital Universitario Central de Asturias) y uno de Barcelona (Hospital Universitari Vall d’Hebron). Se escogió a pacientes con el diagnóstico de SD (n=8) o de EA (n=3) que además hubieran desarrollado crisis epilépticas mioclónicas. La forma más frecuente de acceso de estos pacientes al neurólogo fue a través del servicio de urgencias, donde acudían tras presentar crisis epilépticas. En todos ellos se contó con datos de la historia clínica relacionados con el seguimiento clínico, con estudios de neuroimagen (TC y/o resonancia magnética craneal) y con exploración electroencefalográfica (EEG).
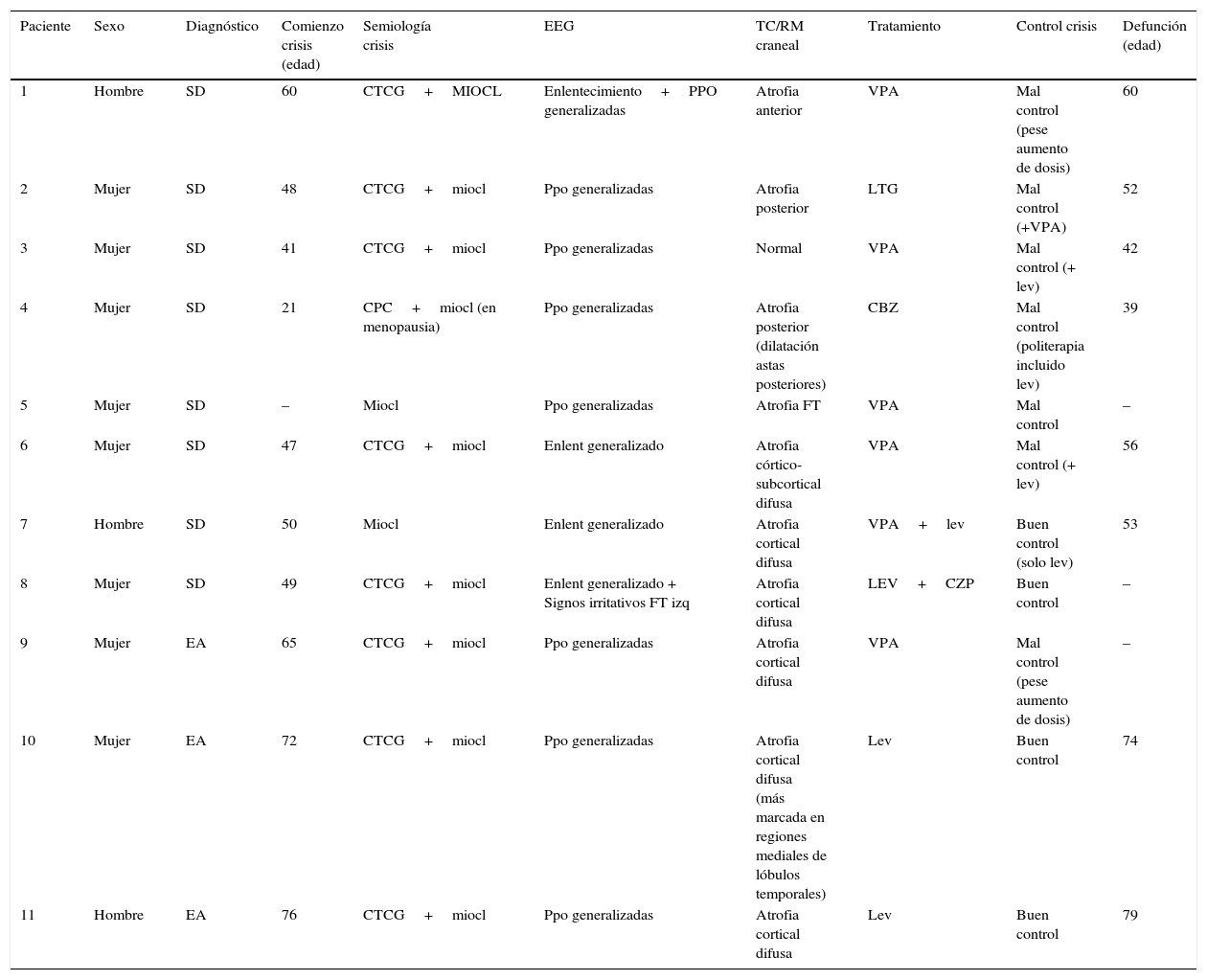
ResultadosLas características demográficas, clínicas y exploraciones complementarias de los pacientes se encuentran resumidas en la tabla 1. En el grupo de pacientes con SD, el rango de edad oscila entre los 21 y 65 años, siendo el 75% mujeres. La edad media (x¯) de inicio de las crisis fue 45,1 años. Tres de ellos (pacientes 5, 6 y 8) ya eran dependientes para las actividades básicas de la vida diaria antes de iniciar la clínica epiléptica. De estos, 2 casos (pacientes 5 y 8) presentaban una historia de deterioro cognitivo sobre su déficit intelectual basal, diagnosticándose de posible EA en paciente con SD.
Características demográficas, clínicas y exploraciones complementarias de los pacientes
| Paciente | Sexo | Diagnóstico | Comienzo crisis (edad) | Semiología crisis | EEG | TC/RM craneal | Tratamiento | Control crisis | Defunción (edad) |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Hombre | SD | 60 | CTCG+MIOCL | Enlentecimiento+PPO generalizadas | Atrofia anterior | VPA | Mal control (pese aumento de dosis) | 60 |
| 2 | Mujer | SD | 48 | CTCG+miocl | Ppo generalizadas | Atrofia posterior | LTG | Mal control (+VPA) | 52 |
| 3 | Mujer | SD | 41 | CTCG+miocl | Ppo generalizadas | Normal | VPA | Mal control (+ lev) | 42 |
| 4 | Mujer | SD | 21 | CPC+miocl (en menopausia) | Ppo generalizadas | Atrofia posterior (dilatación astas posteriores) | CBZ | Mal control (politerapia incluido lev) | 39 |
| 5 | Mujer | SD | – | Miocl | Ppo generalizadas | Atrofia FT | VPA | Mal control | – |
| 6 | Mujer | SD | 47 | CTCG+miocl | Enlent generalizado | Atrofia córtico-subcortical difusa | VPA | Mal control (+ lev) | 56 |
| 7 | Hombre | SD | 50 | Miocl | Enlent generalizado | Atrofia cortical difusa | VPA+lev | Buen control (solo lev) | 53 |
| 8 | Mujer | SD | 49 | CTCG+miocl | Enlent generalizado + Signos irritativos FT izq | Atrofia cortical difusa | LEV+CZP | Buen control | – |
| 9 | Mujer | EA | 65 | CTCG+miocl | Ppo generalizadas | Atrofia cortical difusa | VPA | Mal control (pese aumento de dosis) | – |
| 10 | Mujer | EA | 72 | CTCG+miocl | Ppo generalizadas | Atrofia cortical difusa (más marcada en regiones mediales de lóbulos temporales) | Lev | Buen control | 74 |
| 11 | Hombre | EA | 76 | CTCG+miocl | Ppo generalizadas | Atrofia cortical difusa | Lev | Buen control | 79 |
-: dato no disponible; CBZ: carbamazepina; CPC: crisis parciales complejas; CTCG: crisis tónico-clónicas generalizadas; CZP: clonazepam; EA: enfermedad de Alzheimer; F: frontal; FT: frontotemporal; LEV: levetiracetam; LTG: lamotrigina; MIOCL: mioclonías; PPO: polipunta-onda; SD: síndrome de Down; VPA: ácido valproico.
Respecto a los pacientes con EA (pacientes 9, 10 y 11), las edades en el momento de recogida de datos eran 68, 74 y 78 años; siendo 2 mujeres y un hombre. Su genotipo ApoE era 3,3, 3,4 y 4,4, respectivamente. Se excluyeron mutaciones en los genes PS1, PS2 y APP. Los 3 eran ya dependientes para realizar actividades instrumentales antes de la aparición de la epilepsia con un GDS de 4 en los 3 casos.
La semiología de las crisis consistía fundamentalmente en crisis tónico-clónicas y mioclonías, aunque una paciente presentaba crisis parciales complejas que comenzaron años antes que las mioclonías. Las pacientes 4 y 6 presentaban una historia de epilepsia previamente al inicio de las mioclonías; si bien con el inicio de estas —tras la menopausia en la paciente 4 y a los 53 años en la paciente 6—, comenzó un deterioro cognitivo y desarrollaron refractariedad al tratamiento antiepiléptico.
La TC craneal mostró atrofia cerebral de predominio cortical en todos los pacientes. Se realizó EEG, observándose en todos ellos un enlentecimiento de la actividad de fondo y además en 8 pacientes se evidenciaron paroxismos generalizados de polipunta-onda.
Se consiguió una disminución en el número de crisis epilépticas con menor riesgo de caídas en el 36,4% de los pacientes, siendo el valproato (n=6) y el levetiracetam (n=4) los antiepilépticos más usados. En los pacientes con buena respuesta a medicación antiepiléptica, la monoterapia con levetiracetam fue suficiente (n=4). La dosis de levetiracetam en nuestros pacientes se encontraba entre 500 y 2.000mg al día. Con el tratamiento se consiguió facilitar el cuidado de los pacientes.
Al examinar la evolución, en todos los casos se evidenció un importante empeoramiento cognitivo y funcional en el contexto de las crisis epilépticas. Los 3 pacientes con EA pasaron de GDS 4 a GDS 5. Siete pacientes fallecieron dentro de los 5 primeros años desde el inicio de las mioclonías, con una media de supervivencia de 3 años (tabla 1), siendo la principal causa de defunción las complicaciones infecciosas, de las cuales, la más frecuente fue la infección respiratoria por broncoaspiración.
DiscusiónEn nuestra serie, la aparición tardía de epilepsia constituye un punto de inflexión en la evolución de los pacientes, objetivándose a partir de la misma el inicio o la agudización del deterioro de las funciones cognitivas y de la capacidad funcional. Las características clínicas de nuestros pacientes son similares a las de otras series de la literatura6,7. En lo referente al tratamiento, se puede conseguir una disminución del número de crisis epilépticas en muchos pacientes con la utilización de nuevos antiepilépticos, como el levetiracetam. Este último es el único que ha demostrado una disminución en la frecuencia de crisis epilépticas en modelos animales de ratones con PPA humano sobreexpresado con posible mejoría a nivel conductual y cognitivo8. Respecto al valproato, su papel en la EA sigue siendo controvertido, ya que pese a que podría tener efectos neurotóxicos con una mayor aceleración en la pérdida de volumen cerebral, esto no se acompaña de alteraciones relevantes en las escalas de valoración cognitiva en pacientes con EA9. No obstante, sería razonable evitar el valproato en pacientes de edad avanzada debido a sus efectos adversos10. En este apartado, es importante tener en cuenta que algunos antiepilépticos que actúan sobre el canal de sodio, como la fenitoína o la carbamazepina, podrían producir un empeoramiento de las crisis epilépticas mioclónicas11.
Como ya se ha mencionado previamente en todos las personas con SD y deterioro cognitivo leve hay cambios anatomopatológicos compatibles con EA; sin embargo, no todos los pacientes desarrollan clínica de demencia2. Este hecho ha provocado en los últimos años un interés creciente sobre la fisiopatología de la enfermedad en esta población. En este sentido, en los criterios neuropatológicos del 2012 de la National Institute on Aging y la Alzheimer's Association12, ya se establece claramente que no es necesario tener establecido un diagnóstico clínico de EA para poder realizar un diagnóstico neuropatológico de esta enfermedad. Este criterio sigue la línea comenzada en 2011 con los nuevos criterios diagnósticos de la EA13, en los cuales se introducía la idea de biomarcadores que tuviesen una correlación con las lesiones neuropatológicas con el fin de facilitar un posible diagnóstico precoz.
En un principio, se pensaba que la epilepsia podría ser secundaria a la neurodegeneración producida por el depósito de proteína Aβ. No obstante, en los últimos años se ha descrito que las alteraciones de los neurotransmisores en el balance excitatorio-inhibitorio no solo provocarían crisis epilépticas, sino que podrían contribuir al deterioro cognitivo14 y es posible que el péptido Aβ produzca disfunción sináptica, la cual ocurriría antes que la pérdida neuronal15.
En la EA es bien conocida la degeneración de los sistemas colinérgicos y glutamatérgicos. No obstante, en contraste, el sistema del ácido gamma-aminobutírico parece que se encuentra preservado16. En este sentido, se ha descrito que un canal de Na+ (Nav1.1) podría tener relevancia tanto en la epileptogénesis como el deterioro cognitivo en ratones PPA15.
También la proteína tau —proteína asociada a los microtúbulos axonales, cuya principal función es estabilizarlos— podría desempeñar un papel importante en la epileptogénesis, ya que en ratones con demencia tipo Alzheimer se reduce la intensidad y la frecuencia de las crisis epilépticas al disminuir la proteína tau17. Por último, en la EA autosómica dominante en relación con la mutación del gen de la presenilina 1 (PSEN1) puede haber una epilepsia de inicio tardío18, si bien la etiopatogenia no está estudiada ante la amplia variabilidad de fenotipos en esta mutación.
Actualmente, se están realizando estudios en modelos animales con inhibidores de secretasas, inmunoterapia, antagonistas de mGluR5 e inhibidores de la transcripción del gen APP. A pesar de ello, estos fármacos todavía no se usan en el tratamiento de forma habitual y tampoco por el momento hay biomarcadores que estén disponibles en la práctica habitual.
ConclusionesLos importantes avances en la atención médica de los últimos años han aumentado mucho la esperanza de vida de los pacientes con SD, permitiendo que en la actualidad muchos superen la barrera de los 50 años. Además, ha habido un gran cambio en la mentalidad de la sociedad buscando que estos pacientes se integren en la misma y realicen una vida lo más activa posible.
En este contexto, la labor del neurólogo es fundamental en la detección de un empeoramiento de las actividades diarias habituales o en la aparición de crisis epilépticas. Un tratamiento antiepiléptico adecuado puede controlar las crisis epilépticas, facilitando el cuidado de estos pacientes y mejorando el pronóstico a corto plazo.
Actualmente, no hay datos objetivos a nivel etiopatogénico que permitan definir a LOMEDS como una entidad nosológica independiente. Pese a que cuando aparece se asocia a un importante y rápido empeoramiento de las funciones cognitivas; no se puede descartar que en realidad se trate de un proceso continuo más larvado, que se haría más evidente en los últimos estadios de la enfermedad.
Por otra parte, es innegable el interés teórico de este proceso y de la epileptogénesis en la EA, que ha permitido ampliar los conocimientos de la enfermedad y desarrollar nuevas estrategias terapéuticas. Los pacientes con SD, debido a la gran variabilidad fenotípica que presentan y a una mayor incidencia de epilepsia que en la población general, han facilitado importantes avances científicos en este campo.
Conflicto de interesesDeclaración de conflictos de intereses: el autor primer firmante del manuscrito de referencia, en su nombre y en el de todos los autores firmantes, declara que no existe ningún potencial conflicto de interés relacionado con el artículo.
Tres pacientes de esta serie fueron usados en la comunicación oral «Epilepsia mioclónica de comienzo tardío en el síndrome de Down», en la LXIV Reunión Anual de la SEN en Barcelona, en noviembre del 2012..

