



Patients with Down syndrome (DS) who exhibit Alzheimer disease (AD) are associated with age. Both diseases with a common neuropathological basis have been associated with late-onset myoclonic epilepsy (LOMEDS). This entity presents electroencephalogram features as generalised polyspike-wave discharges.
MethodWe present a series of 11 patients with the diagnosis of DS or AD who developed myoclonic seizures or generalised tonic–clonic seizures. In all cases, clinical and neuroimaging studies and polygraph EEG monitoring was performed.
ResultsIn all cases, cognitive impairment progressed quickly after the onset of epilepsy causing an increase in the degree of dependence. The most common finding in the EEG was a slowing of brain activity with theta and delta rhythms, plus intercritical generalised polyspike-waves were objectified in eight patients. In neuroimaging studies was found cerebral cortical atrophy. The most effective drug in this series was the levetiracetam.
ConclusionsThe association of generalised epilepsy with elderly DS represents an epiphenomenon in evolution which is associated with a progressive deterioration of cognitive and motor functions. This epilepsy has some electroclinical characteristics and behaves as progressive myoclonic epilepsy, which is probably related to the structural changes that characterise the evolutionary similarity of DS with AD. Recognition of this syndrome is important, since it has prognostic implications and requires proper treatment.
Los pacientes con síndrome de Down (SD) presentan una demencia tipo Alzheimer (EA) asociada a la edad. Ambas patologías, con una base neuropatológica común, han sido asociadas a la epilepsia mioclónica de inicio tardío (LOMEDS). Esta entidad presenta alteraciones electroencefalográficas características en forma de descargas generalizadas de polipunta-onda.
MétodoPresentamos una serie de 11 pacientes con el diagnóstico de SD o EA que desarrollaron crisis epilépticas mioclónicas o tónico-clónicas generalizadas. En todos ellos, se realizó un seguimiento clínico y estudios de neuroimagen y poligrafía EEG.
ResultadosEn todos los casos, el deterioro cognitivo avanzó rápidamente tras el comienzo de la epilepsia, produciendo un incremento en el grado de dependencia. El hallazgo más común en el EEG fue un enlentecimiento de la actividad cerebral con ritmos theta y delta; además, en 8 pacientes se objetivaron descargas intercríticas generalizadas de polipunta-onda. En los estudios de neuroimagen se encontró atrofia cerebral cortical. El fármaco más eficaz en esta serie fue el levetiracetam.
ConclusionesLa asociación de epilepsia generalizada al SD de edad avanzada supone un epifenómeno en la evolución que marca un agravamiento rápidamente progresivo de las funciones cognitivas y motoras. Presenta unas características electroclínicas bien definidas y se comporta como una epilepsia mioclónica progresiva, que probablemente se relaciona con los cambios estructurales que caracterizan el parecido evolutivo del SD con la enfermedad de Alzheimer. El reconocimiento de este síndrome es importante, dado que tiene repercusiones pronósticas y requiere un tratamiento adecuado.
The prevalence of epileptic seizures in patients with Alzheimer disease (AD) is higher than in the normal population.1 Prevalence of epilepsy in patients with Down syndrome (DS) increases with age and reaches 46% in patients older than 50.2 As in the normal population, incidence of focal epilepsy of structural causes increases with age in individuals with DS.
Anatomical pathology studies have described changes compatible with AD in the brains of DS patients who had developed dementia or mild cognitive impairment.3 Trisomy 21 plays a significant role in the pathogenesis of these changes since chromosome 21 contains the gene coding for amyloid precursor protein (APP) — which increases production of amyloid protein, and the gene coding for the beta-site amyloid precursor protein cleaving enzyme 2 (BACE 2) — which generates the amyloid peptides making up senile plaques. Such other factors as oestrogens, APOE alleles, and prion protein (PRNP) gene polymorphisms are also involved.
Epilepsy was believed to be a typically late-onset phenomenon in the course of AD. However, Lozsadi and Larner4 report that up to 6.8% of the patients with a new diagnosis of clinically probable AD will present epilepsy and need antiepileptic treatment. In 3.4% of these cases, epileptic seizure onset coincides with onset of cognitive impairment.
Myoclonic seizures have been described in DS patients in the context of AD dementia; these seizures are progressive and coincide with a significant acceleration of cognitive decline and increased level of dependence. The entity was first described in 1994 by Genton and Paglia.5 It is characterised by myoclonus occurring predominantly in the morning hours and affecting the upper limbs most of all. They are associated with generalised discharges of polyspike-wave complexes in EEG studies.
Our study describes a series of patients with DS or AD and myoclonus and generalised tonic–clonic seizures in order to better understand these symptoms and improve management of these patients.
MethodThis descriptive study is based on a retrospective review of a series of patients from 3 hospitals in Asturias (Hospital Alvarez Buylla, Hospital Valle del Nalón, and Hospital Universitario Central de Asturias) and a hospital in Barcelona (Hospital Universitari Vall d’Hebron). We selected patients diagnosed with DS (n=8) or AD (n=3) who had also developed myoclonic seizures. The most common means of access to neurologists for these patients was through the emergency department after suffering an epileptic seizure. We obtained each patient's medical data related to clinical follow-up, neuroimaging studies (CT and/or cranial MRI), and EEG study.
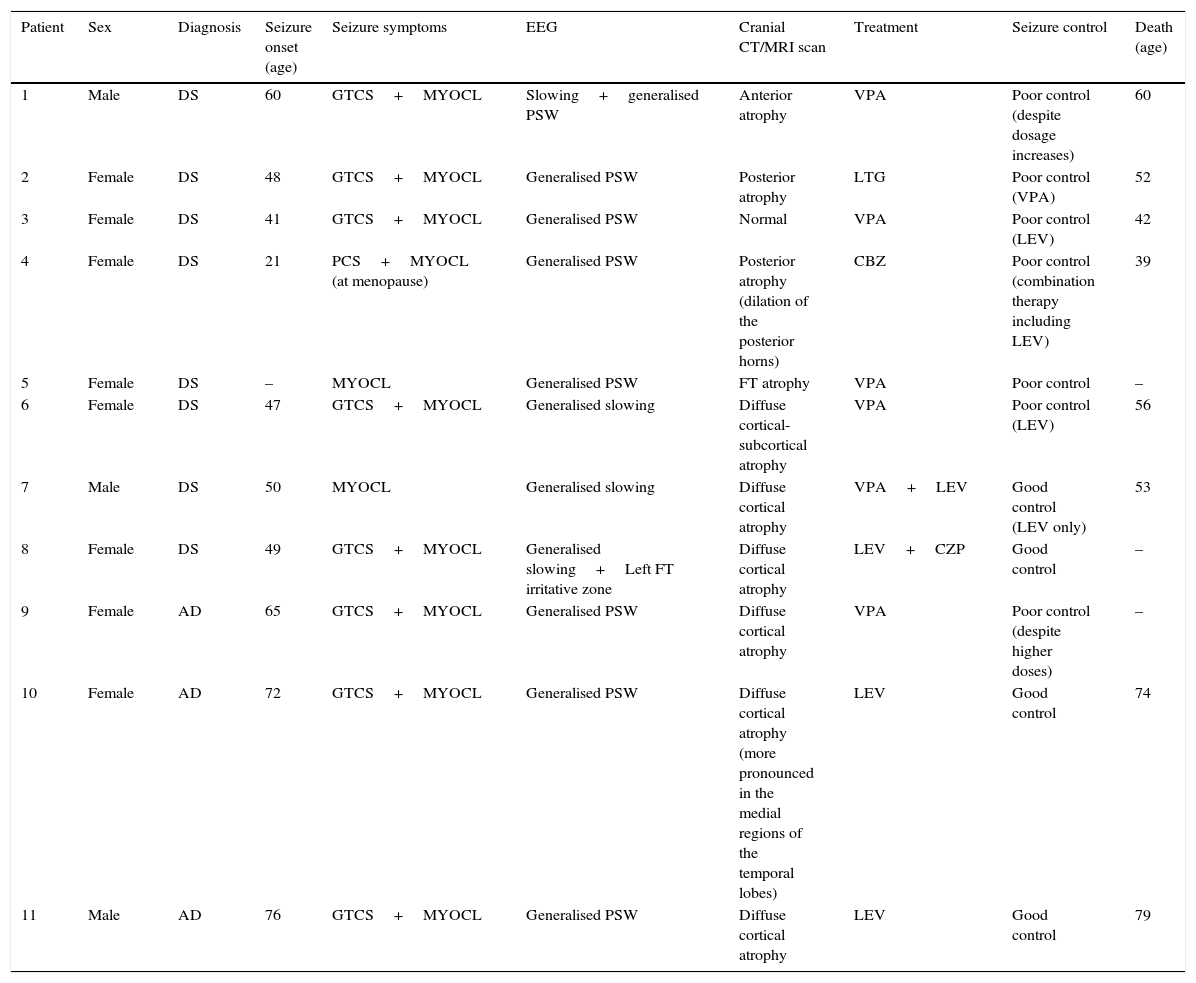
ResultsPatients’ demographic and clinical characteristics as well as data from complementary tests are included in Table 1. In the DS patient group, ages ranged from 21 to 65 years; 75% were women. Mean age (x¯) at seizure onset was 45.1 years. Three patients (patients 5, 6, and 8) were already dependent for activities of daily living before epileptic symptoms manifested. Two of them (5 and 8) had a history of cognitive decline in addition to their intellectual deficits (DS), and they were diagnosed with probable AD.
Patients’ demographic and clinical characteristics and complementary test results.
| Patient | Sex | Diagnosis | Seizure onset (age) | Seizure symptoms | EEG | Cranial CT/MRI scan | Treatment | Seizure control | Death (age) |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Male | DS | 60 | GTCS+MYOCL | Slowing+generalised PSW | Anterior atrophy | VPA | Poor control (despite dosage increases) | 60 |
| 2 | Female | DS | 48 | GTCS+MYOCL | Generalised PSW | Posterior atrophy | LTG | Poor control (VPA) | 52 |
| 3 | Female | DS | 41 | GTCS+MYOCL | Generalised PSW | Normal | VPA | Poor control (LEV) | 42 |
| 4 | Female | DS | 21 | PCS+MYOCL (at menopause) | Generalised PSW | Posterior atrophy (dilation of the posterior horns) | CBZ | Poor control (combination therapy including LEV) | 39 |
| 5 | Female | DS | – | MYOCL | Generalised PSW | FT atrophy | VPA | Poor control | – |
| 6 | Female | DS | 47 | GTCS+MYOCL | Generalised slowing | Diffuse cortical-subcortical atrophy | VPA | Poor control (LEV) | 56 |
| 7 | Male | DS | 50 | MYOCL | Generalised slowing | Diffuse cortical atrophy | VPA+LEV | Good control (LEV only) | 53 |
| 8 | Female | DS | 49 | GTCS+MYOCL | Generalised slowing+Left FT irritative zone | Diffuse cortical atrophy | LEV+CZP | Good control | – |
| 9 | Female | AD | 65 | GTCS+MYOCL | Generalised PSW | Diffuse cortical atrophy | VPA | Poor control (despite higher doses) | – |
| 10 | Female | AD | 72 | GTCS+MYOCL | Generalised PSW | Diffuse cortical atrophy (more pronounced in the medial regions of the temporal lobes) | LEV | Good control | 74 |
| 11 | Male | AD | 76 | GTCS+MYOCL | Generalised PSW | Diffuse cortical atrophy | LEV | Good control | 79 |
–: not available; CBZ: carbamazepine; CPS: complex partial seizures; GTCS: generalised tonic–clonic seizures; CZP: clonazepam; AD: Alzheimer disease; F: frontal; FT: frontotemporal; LEV: levetiracetam; LTG: lamotrigine; MYOCL: myoclonus; PSW: poly-spike wave; DS: Down syndrome; VPA: valproic acid.
In the AD patient group (patients 9, 10, and 11), ages at time of data collection were 68, 74, and 78; 2 were women. Their APOE genotypes were 3.3, 3.4, and 4.4, respectively. We excluded mutations in PS1, PS2, and APP genes. These 3 patients were already dependent for instrumental activities before epilepsy manifested; GDS was 4 for all 3 cases.
Seizure semiology mainly consisted of tonic–clonic seizures and myoclonus, although one patient initially presented complex partial seizures years before experiencing myoclonus. Patients 4 and 6 had a history of epilepsy before onset of myoclonus; myoclonus (after menopause in patient 4 and at age 53 in patient 6) was accompanied by cognitive impairment, and seizures became refractory to antiepileptic treatment.
Cranial CT scan showed predominantly cortical brain atrophy in all patients. EEGs revealed a slowing of background activity in all patients. In addition, we observed generalised polyspike-wave paroxysms in 8 patients.
Treatment was successful in decreasing the number of seizures and the risk of falling in 36.4% of the patients; the most widely used antiepileptic drugs were valproate (n=6) and levetiracetam (n=4). In patients with a good response to antiepileptic treatment, monotherapy with levetiracetam was sufficient for seizure control (n=4). Doses of levetiracetam in our patients were between 500 and 2000mg/day. Caring for patients became easier with seizure treatment.
Regarding progression, significant cognitive and functional decline accompanied epileptic seizures in all patients. The 3 patients with AD obtained GDS scores of 5 (previously 4). Seven patients died within 5 years of onset of myoclonus, with a mean survival of 3 years (Table 1). The main cause of death was infectious complications, with the most frequent being aspiration-related pulmonary infection.
DiscussionIn our series, late-onset myoclonic epilepsy (LOMEDS) constitutes a turning point in the course of the disease; impairment of cognitive functions and functional capacity starts or exacerbates with epilepsy onset. Clinical characteristics of our patients are similar to those in other published series.6,7 Regarding treatment, the number of epileptic seizures can be reduced in many patients by using new antiepileptic drugs, such as levetiracetam. This is the only drug demonstrated to decrease seizure frequency in mouse models with overexpression of human APP, with the possibility of improvement at the behavioural and cognitive levels.8 The role of valproate in AD remains controversial; despite having potentially neurotoxic effects and accelerating brain volume loss, it is not associated with significant changes in AD patients’ scores on cognitive assessment scales.9 It would be reasonable to avoid valproate in elderly patients due to its adverse effects.10 It is important to remember that some antiepileptic drugs that act on the sodium channel, such as phenytoin or carbamazepine, could exacerbate myoclonic seizures.11
As mentioned previously, all subjects with DS and mild cognitive impairment show anatomopathological changes compatible with AD, although not all patients develop dementia symptoms.2 This observation has sparked a growing interest in the pathophysiology of AD in this population. In light of this interest, the 2012 neuropathological criteria of the National Institute on Ageing and the Alzheimer's Association12 clearly established that a clinical diagnosis of AD is not required to assign a neuropathological diagnosis of this disease. This criterion is compatible with the new diagnostic criteria for AD established in 2011,13 and which introduced the idea of biomarkers that may be correlated with neuropathological lesions, a situation that would foster the possibility of early diagnosis.
At one time, epilepsy was thought to be secondary to the neurodegeneration caused by deposition of Aβ protein. Nevertheless, in recent years, neurotransmitter alterations in the excitatory-inhibitory balance have been reported not only to provoke epileptic seizures but also to contribute to cognitive impairment.14 In addition, Aβ peptide may cause synaptic dysfunction, which would occur before neuronal loss.15
Degeneration of cholinergic and glutaminergic systems is a well-established process in AD, although the gamma-aminobutyric acid system seems to be preserved.16 Similarly, a Na+ channel (Nav1.1) was found to be potentially relevant for both epileptogenesis and cognitive impairment in APP mice.15
Tau protein (an axonal microtubule-associated protein whose main function is to stabilise microtubules) is another factor that may play a fundamental role in epileptogenesis: mice with AD-type dementia show reduced intensity and frequency of epileptic seizures when tau protein also decreases.17 Lastly, late-onset epilepsy may manifest in autosomal dominant AD related to a presenilin 1 (PSEN1) gene mutation,18 although the aetiopathogenesis has not been studied sufficiently considering the wide variety of phenotypes of this mutation.
Animal model studies featuring secretase inhibitors, immunotherapy, mGluR5 inhibitors, and APP transcription inhibitors are currently underway. However, these drugs are not yet being used as typical treatments, and no biomarkers are available for use in daily practice.
ConclusionsThe past few years have ushered in advances in medical care that have significantly increased the life expectancy of DS patients, many of whom now live longer than 50 years. Furthermore, the mindset of society at large has evolved considerably; patients are now better integrated so that their lives can be as active as possible.
In this context, the work of the neurologist is essential for detecting loss of competence for activities of daily living and increased frequency of epileptic seizures. Appropriate antiepileptic treatment may achieve seizure control, which will facilitate patient care and improve short-term outcomes.
At present, objective aetiopathogenic data is not sufficient to define LOMEDS as an independent nosological entity. When this disease manifests, it is usually accompanied by a marked and rapid cognitive decline; however, this entity may also be a more latent continuous process which becomes more evident in later stages of the disease.
On the other hand, we cannot deny the theoretical relevance of this process and of epileptogenesis in AD; new findings have allowed us to expand our knowledge of the disease and develop new treatment strategies. Studying DS patients, who present a wide variety of phenotypes and a higher incidence of epilepsy than that of the general population, has resulted in important scientific advances in this field.
Conflicts of interestThe lead author of this article, on behalf of himself and of all coauthors, states that there are no potential conflicts of interest related to this study.
Please cite this article as: Aller-Alvarez JS, Menéndez-González M, Ribacoba-Montero R, Salvado M, Vega V, Suárez-Moro R, et al. Epilepsia mioclónica en el síndrome de Down y en la enfermedad de Alzheimer. Neurología. 2017;32:69–73.
Three patients in this series were mentioned in the oral communication “Myoclonic epilepsy in Down syndrome and Alzheimer disease”, presented during the 64th Annual Meeting of the Spanish Society of Neurology (Barcelona), November 2012.
articles

Neurología (English Edition) follows the Recommendations for the Conduct, Reporting, Editing and Publication of Scholarly Work in Medical Journals