





La hemocromatosis hereditaria es el trastorno genético más común en la población caucásica, con una prevalencia estimada de 1/200-1/400 (homocigotos) y de 1/8-1/10 en portadores (heterocigotos). Tiene una transmisión de carácter recesivo y ligado al HLA. La expresión clínica de la enfermedad aparece solo en homocigotos y va depender del momento del diagnóstico y del grado de afectación orgánica.
El caso clínico a continuación presentamos describe el proceso del diagnóstico diferencial realizado en un paciente joven con coxartosis unilateral, y sospecha clínica de hemocromatosis.
El diagnóstico precoz de la hemocromatosis hereditaria en atención primaria, puede disminuir la morbimortalidad de esta enfermedad al detectar homocigotos en edades tempranas.
Hereditary haemochromatosis is the most common genetic disorder in the Caucasian population, with an estimated prevalence of 1/200-1/400 (homozygous) and 1/8-1/10 in carriers (heterozygous). The transmission is recessive and linked to HLA. The clinical expression of disease appears only in homozygous and will depend on the time of diagnosis and the degree of organ involvement.
The clinical case presented below describes the process of differential diagnosis made in a young patient with unilateral coxarthrosis and clinical suspicion of hemochromatosis.
Early diagnosis of hereditary hemochromatosis in primary care can reduce morbidity and mortality of this disease by detecting homozygous at younger ages.
La hemocromatosis es una enfermedad que interfiere con el metabolismo del hierro provocando un aumento de su absorción en la mucosa gastrointestinal y originando un depósito progresivo en las células parenquimatosas de diferentes órganos. El exceso de hierro se acumula inicialmente en el hígado provocando hepatomegalia, posteriormente se pueden afectar otros órganos y desarrollarse con el tiempo diabetes, cambios de pigmentación de la piel, cardiopatía, artritis, atrofia testicular, cirrosis hepática e hipopituitarismo1.
Existen 2 tipos: hemocromatosis hereditaria (HH) y hemocromatosis secundaria (HS) a procesos o situaciones que producen una acumulación de hierro en los órganos: porfiria cutánea tardía, anemia hemolítica, ingestión oral crónica de hierro, alcoholismo crónico y transfusiones sanguíneas crónicas2.
El síntoma más frecuente de la HH es la astenia pero es muy inespecífico. Las manifestaciones clínicas de la enfermedad tienen lugar solo en homocigotos, es variable y depende de 2 factores: heterogeneidad fenotípica y el momento del diagnóstico.
El cribado de la HH se realiza midiendo el grado de saturación de la transferrina en aquellos pacientes con sospecha clínica y analítica de la enfermedad (niveles de hierro y ferritina superiores al rango de la normalidad). El diagnóstico precoz es esencial para detectar homocigotos en edades tempranas, y así prevenir la aparición precoz de manifestaciones clínicas y reducir la mortalidad.
El diagnóstico de confirmación se realiza mediante pruebas genéticas en aquellos pacientes con cribado positivo. Los tests detectan la mutación C282Y del gen HFE asociado a esta enfermedad (83%) y en menor proporción la mutación H63D (5%).
El tratamiento de elección es la depleción de hierro mediante flebotomías. Si el tratamiento se realiza antes del desarrollo de la enfermedad, la expectativa de vida de los pacientes casi se normaliza.
El pronóstico es malo en estadios avanzados con afectación multiorgánica. En estas fases pueden aparecer las complicaciones más graves de la enfermedad: insuficiencia hepática secundaria a cirrosis avanzada y cáncer hepatocelular3.
A continuación presentamos un caso clínico de HH con afectación articular unilateral.
Caso clínicoVarón de 37 años que acudió al centro de salud por dolor localizado en la zona inguinal izquierda de un mes de duración, acompañado de impotencia funcional y que disminuía parcialmente con la toma de ibuprofeno.
Como antecedentes personales se encontraban sedentarismo, obesidad y HTA esencial tratada con 600mg/día de eprosartán.
En la exploración física se obtuvieron los siguientes datos: peso 92kg, IMC (32,5%), PA: 140/85mm Hg, piel y mucosas de coloración e hidratación normales. Sistema neurológico sin alteraciones. Auscultación cardiopulmonar sin hallazgos patológicos. Abdomen globuloso, sin organomegalias a la palpación. Pliegues inguinales y genitales de aspecto y morfología normal. Rotación interna de la cadera izquierda muy dolorosa entre 0-30°. Cadera derecha asintomática. Pulsos distales normales. Sensibilidad, reflejos osteotendinosos y tono muscular en extremidades sin alteraciones.
Se solicitaron radiografías de ambas caderas, analítica (hemograma, VSG, sedimento de orina, bioquímica básica, perfil lipídico, hepático y metabólico) y se pautó naproxeno sódico 550mg/12h para alivio del dolor.
El paciente acudió a los 15 días relatando disminución del dolor pero continuaba con impotencia funcional en el miembro inferior izquierdo. El resultado de las pruebas complementarias reflejaba un hemograma, bioquímica básica, perfil lipídico, perfil hepático y metabólico sin alteraciones excepto niveles de hierro de 205μg/dl (niveles normales 50-150μg/dl). En la radiología se observaba una disminución del espacio interarticular de la cadera izquierda con presencia de osteofitos satélites, y no había afectación osteoarticular en la cadera derecha.
Al reevaluar nuevamente al paciente, este relataba haber estado hace 5 años en consulta de un gastroenterólogo por presentar valores aumentados de ferritina en sangre, pero dejó de acudir a los controles por motivos de trabajo.
Con los nuevos datos de la anamnesis y ante la presencia de artrosis unilateral de cadera en un paciente joven, sin antecedentes traumáticos ni infecciosos previos, se procedió a realizar una petición de pruebas complementarias basadas en un diagnóstico diferencial de causas de artrosis secundarias, incluyendo la hemocromatosis.
Se solicitó: capacidad de saturación de transferrina, hierro sérico y ferritina, para descartar hemocromatosis y se amplío el estudio con coagulación, ecografía hepática, estudio virológico (VHB, VHC y VIH), inmunoglobulinas, ANA, ANOES, α-fetoproteína, TSH, PTH y FR, para detectar otras causas de artrosis secundaria. Se mantuvo el tratamiento con AINE hasta nueva valoración.
Las pruebas complementarias constataban concentraciones de hierro de 201μg/dl (niveles normales 50-150μg/dl); transferrina: 233mg/dl (niveles normales 175-400mg/dl); ferritina: 210ng/ml (niveles normales 30-300ng/ml), y saturación de transferrina: 80% (niveles normales: 20-50%). El hemograma, bioquímica básica, perfil hepático, perfil metabólico, perfil lipídico, coagulación, inmunología y α-fetoproteína no presentaban alteraciones.
La ecografía abdominal detectó una esteatosis hepática con un bazo en el límite de tamaño de la normalidad (fig. 1).

Se decidió inicialmente derivar al paciente a consultas de gastroenterología por presentar alta sospecha clínica y cribado positivo para hemocromatosis.
El resultado del estudio del especialista concluyó que el paciente era homocigoto para la mutación C282Y del gen asociado a la hemocromatosis y se amplió el estudio genético a familiares de primer grado. Le pautaron como tratamiento sangrías cada 15 días durante 3 meses y al finalizar las sangrías el nivel de ferritina descendió a 16ng/ml, el hierro a 26 μg/ml y la saturación de transferrina al 23%, pero el paciente continuaba con dolor moderado e impotencia funcional.
Desde la consulta de atención primaria y con el consenso del paciente se realizó una derivación a traumatología para valorar la indicación de prótesis de cadera y ampliar el estudio de causas de coxartrosis secundaria. Se aconsejó la realización de biopsia hepática pero el paciente la rechazó.
En la actualidad el paciente está en lista de espera para prótesis de cadera y se han descartado otras enfermedades causantes de artrosis secundaria.
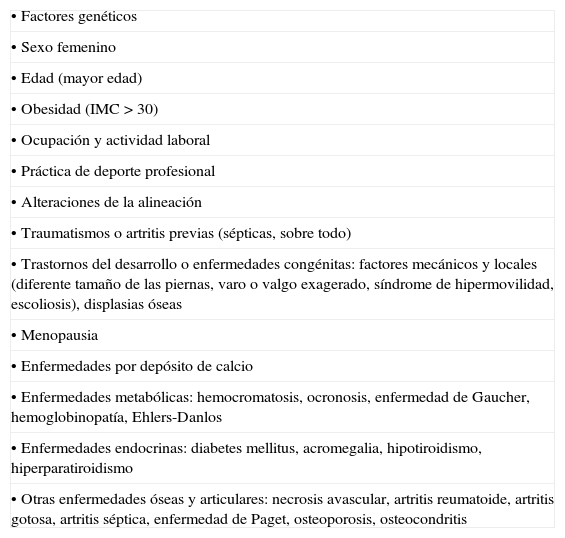
DiscusiónLa presencia de artrosis unilateral de cadera en un paciente joven, sin antecedentes traumáticos ni infecciosos previos, obliga a realizar un diagnóstico diferencial de artrosis secundaria. Una de las posibles causas de artrosis secundaria es la hemocromatosis (tabla 1).
Causas de artrosis
| • Factores genéticos |
| • Sexo femenino |
| • Edad (mayor edad) |
| • Obesidad (IMC > 30) |
| • Ocupación y actividad laboral |
| • Práctica de deporte profesional |
| • Alteraciones de la alineación |
| • Traumatismos o artritis previas (sépticas, sobre todo) |
| • Trastornos del desarrollo o enfermedades congénitas: factores mecánicos y locales (diferente tamaño de las piernas, varo o valgo exagerado, síndrome de hipermovilidad, escoliosis), displasias óseas |
| • Menopausia |
| • Enfermedades por depósito de calcio |
| • Enfermedades metabólicas: hemocromatosis, ocronosis, enfermedad de Gaucher, hemoglobinopatía, Ehlers-Danlos |
| • Enfermedades endocrinas: diabetes mellitus, acromegalia, hipotiroidismo, hiperparatiroidismo |
| • Otras enfermedades óseas y articulares: necrosis avascular, artritis reumatoide, artritis gotosa, artritis séptica, enfermedad de Paget, osteoporosis, osteocondritis |
Fuente: Miguel de E. Clínica y tratamiento de la artrosis periférica. En: Manual SER de las enfermedades reumáticas. 3.a edition. Madrid: Editorial Médica Panamericana; 2000. p. 476-82.
La artropatía aparece entre un 25-50% de los pacientes, generalmente después de los 50 años, aunque también puede aparecer antes, incluso ser la manifestación inicial o mucho tiempo después del tratamiento. Las primeras articulaciones en afectarse son las metacarpofalángicas; sobre todo la segunda y la tercera. A continuación, puede aparecer poliartritis progresiva que afecta a muñecas, caderas, tobillos y rodillas. Las manifestaciones articulares son las únicas que no guardan relación proporcional con el grado de acumulación de hierro en tejidos; y son debidas en su totalidad a la HH. Un aspecto negativo de la artropatía es su tendencia a progresar a pesar de los tratamientos con sangrías; hecho que se observaba en el paciente del caso clínico.
Al tratarse de una enfermedad hereditaria relativamente frecuente, los protocolos de cribado son prioritarios. El diagnóstico precoz adelanta la intervención terapéutica y evita las complicaciones. La saturación de transferrina se recomienda como cribado en hombres y mujeres entre 20-30 años con alteraciones en los parámetros bioquímicos del hierro (ferritina y sideremia). La saturación de tranferrina supera en especificidad y precocidad de aparición de la HH al incremento de los niveles de ferritina. Una ferritinemia elevada solo orienta hacia un aumento de los depósitos de hierro de origen o no hemocromatósico y obliga a la realización de un estudio clínico etiológico. Niveles>a 55% en mujeres y>a 60% en hombres son indicativos de enfermedad4. En estudios poblacionales la saturación de transferrina tiene mejor coste beneficio que las pruebas genéticas para realizar el despistaje o cribado de la enfermedad4.
Las pruebas genéticas se indican solo cuando el cribado ha sido positivo y tenemos que confirmar el diagnóstico de HH5. La mutación C282Y en el cromosoma 6 del gen HFE (83%), y rara vez la H63D, son diagnósticas de enfermedad. La presencia de un portador homocigoto no implica obligatoriamente que éste vaya a presentar enfermedad clínicamente manifiesta, aunque su probabilidad es alta. Los portadores heterocigóticos de la mutación (5-10% de la población) pueden presentar incrementos de los marcadores bioquímicos, pero sin traducción clínica similar a la HH homocigótica.
La biopsia hepática es esencial para realizar un pronóstico de la enfermedad, ya que las pruebas genéticas no proporcionan información acerca del grado de depósito del hierro, ni del daño en orgánico. Se considera excepción para su realización aquellos sujetos jóvenes diagnosticados por pruebas genéticas o estudio familiar y con aumento de los parámetros bioquímicos del hierro, pues en ellos podría iniciarse un tratamiento precoz por la poca probabilidad de presencia de cirrosis y fibrosis (hecho que se consideró en el paciente del caso clínico). Las muestras de biopsia junto con las pruebas genéticas confirman el diagnóstico definitivo. Un índice hepático en (mmol/g) de hierro>2 en la biopsia puede considerarse diagnóstico, aunque no es igual en varones que en mujeres. Si el índice hepático de hierro es<1,5 se excluye la HH homocigótica. Valores entre 1,5-2 deben ser interpretados con precaución y se acepta que valores de índice hepático de hierro >5 en varones y >3 en mujeres corresponden a HH. La biopsia se recomienda utilizarla únicamente cuando se sospecha cirrosis o fibrosis, predecible por ferritina > 1.000mg/l, aumento de transaminasas y/o hepatomegalia6.
Para completar el diagnóstico se puede realizar: tomografía computarizada, ecografía, niveles de alfafetoproteínas, pruebas de función hepática, tamizaje para hepatitis y ECG.
El objetivo del tratamiento de la HH es la depleción de hierro en homocigotos antes del desarrollo de la enfermedad. Las flebotomías constituyen el tratamiento de elección, ya que su realización es simple, son bien toleradas y muy eficaces. Un programa de flebotomías regulares en un estadio premórbido conlleva una expectativa de vida normal para el paciente, evita la aparición de manifestaciones clínicas y las complicaciones de la enfermedad7. La extracción se realiza semanalmente hasta que se alcanzan valores de ferritina <30mg/l y una saturación de transferrina <20%; después la frecuencia se ajusta según estos parámetros. Las sangrías mejoran la función ventricular izquierda y aumentan la supervivencia en pacientes con miocardiopatía dilatada. En pacientes cirróticos la depleción de hierro disminuye el grado de hipertensión portal, pero existe un mayor riesgo de carcinoma hepatocelular aunque se haya eliminado el exceso de hierro durante años. El tratamiento tiene poco efecto cuando existe afectación hipofisaria y diabetes secundaria.
La dieta pobre en hierro no previene, ni controla la hemocromatosis pero se debe evitar el consumo de alimentos que aumenten la sobrecarga existente: alcohol, moluscos (pueden comerse cocidos), cereales que contienen hierro y sobre todo suplementos vitamínicos con hierro.
La pérdida del deseo sexual y el cambio de las características sexuales secundarias mejoran con una terapia de testosterona.
Los quelantes del hierro se utilizan excepcionalmente en pacientes que tengan contraindicación o complicación secundaria a las flebotomías: anemia grave, hipoalbuminemia grave y cardiopatía descompensada6.
Los resultados del trasplante hepático no son tan favorables como en otras enfermedades crónicas del hígado, ya que estos pacientes suelen presentar afectación multiorgánica.
El pronóstico es grave en estadios muy avanzados cuando existe afectación multiorgánica y complicaciones (insuficiencia hepática y cáncer de hígado). El carcinoma hepatocelular causa más del 50% de la mortalidad en estos pacientes.
Los heterocigotos solo desarrollan sobrecarga significativa cuando coexisten otras enfermedades o situaciones que afectan al metabolismo del hierro: β-talasemia heterocigótica, esferocitosis hereditaria, porfiria cutánea tarda, transfusiones crónicas en pacientes con anemias, etc.
Responsabilidades éticasProtección de personas y animales. Los autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datos. Los autores declaran que en este artículo no aparecen datos de pacientes.
Derecho a la privacidad y consentimiento informado. Los autores declaran que en este artículo no aparecen datos de pacientes.
