




En los últimos años se han producido grandes avances en el conocimiento de la patogenia del cáncer colorrectal, que han llevado a la obtención de nuevos fármacos para el tratamiento de esta enfermedad. Asimismo, se dispone de marcadores moleculares que han demostrado su capacidad en la predicción del pronóstico tumoral. Sin embargo, las decisiones acerca del tratamiento adyuvante en el cancer colorrectal continúan tomándose en función, únicamente, del estadio histológico. Diferentes marcadores que pueden ser detectados en el tejido tumoral han mostrado, además, un valor en la predicción de la respuesta a los fármacos antineoplásicos. En este artículo se revisa la posible utilidad clínica de estos marcadores y su papel en la toma de decisions en el tratamiento de los pacientes con cáncer colorrectal.
In the last few years, major advances have been produced in knowledge of the pathogenesis of colorectal cancer, which have led to the development of new drugs for the treatment of this disease. Likewise, molecular markers have been identified that are useful in prognosis. However, decisions on adjuvant therapy in colorectal cancer continue to be based exclusively on histological stage. Distinct markers that can be detected in tumoral tissue may be useful in predicting response to antineoplastic drugs. The present article reviews the possible clinical utility of these markers and their role in decision-making in the treatment of patients with colorectal cancer.
El cáncer colorrectal (CCR) es la segunda neoplasia más frecuente en España. Su incidencia anual se estima en 25.000 casos, y es la responsable del 3% de las muertes en nuestro país1. En los últimos años estamos asistiendo a un notable incremento en las alternativas terapéuticas en pacientes con CCR, con esquemas de quimioterapia adyuvante cada vez más eficaces. La necesidad de tratamiento quimioterápico en un gran número de pacientes, las diferentes respuestas a éstos y el gran número de opciones terapéuticas disponibles hacen necesario el desarrollo de parámetros predictores de respuesta al tratamiento, que permitan individualizarlo con el fin de obtener un mayor rendimiento. En los últimos años se han obtenido grandes avances en el conocimiento de los mecanismos carcinogénicos y de la farmacogenética del CCR. Asimismo, importantes esfuerzos en investigación traslacional han permitido el desarrollo de técnicas para la detección de marcadores moleculares capaces de predecir la respuesta a los quimioterápicos utilizados en el tratamiento del CCR, y muchas de estas técnicas son en este momento accesibles para la mayoría de laboratorios hospitalarios asistenciales. A pesar de todo, la mayoría de estos predictores de respuesta al tratamiento no son utilizados en la práctica clínica habitual, y la aplicación de tratamientos adyuvantes en el CCR está todavía regida por parámetros clinicopatológicos, como el estadio TNM. Se conoce un gran número de marcadores moleculares con potencial capacidad predictiva in vitro de la respuesta a la quimioterapia, pero presentan limitaciones en su reproducibilidad in vivo. Aunque actualmente estos marcadores no priman en la elección del tratamiento quimioterápico, hay evidencias suficientes en algunos de ellos para poder, al menos, colaborar en la orientación y en la individualización de este tratamiento.
El objetivo de esta revisión es mostrar, de manera concisa, los principales marcadores moleculares capaces de predecir la respuesta a los fármacos quimioterápicos utilizados en la práctica clínica en el CCR.
TRATAMIENTO QUIMIOTERÁPICO EN EL CÁNCER COLORRECTALEl tratamiento quimioterápico adyuvante en el CCR sólo debe aplicarse cuando se obtenga un beneficio sobre la supervivencia o la calidad de vida. En tumores localizados, en estadio I, la cirugía se considera curativa y, por tanto, estos pacientes no son candidatos a tratamiento quimioterápico adyuvante2. En cambio, desde principios de los años noventa se sabe que en los tumores con afectación ganglionar, en estadio III, el tratamiento con quimioterapia adyuvante con 5-fluorouracilo (5-FU) modulado con ácido folínico o leucovorín, consigue disminuir de manera significativa el riesgo de recurrencia y la mortalidad3. Recientemente, se ha demostrado que la adición al 5-FU de oxaliplatino mejora los datos de supervivencia libre de enfermedad en estos pacientes, y esta combinación se ha convertido, hoy en día, en el estándar de tratamiento para pacientes con CCR en estadio III4. Actualmente, todavía hay dudas acerca de la eficacia de la quimioterapia adyuvante en pacientes con CCR en estadio II, y por este motivo sólo se indica tratamiento a los considerados de alto riesgo, que son los casos con resección ganglionar regional incompleta, las lesiones T4, la presentación como perforación u obstrucción intestinal, la presencia de invasión linfática, vascular o perineural y los tumores con histología escasamente diferenciada5,6. También son susceptibles de quimioterapia los cánceres en estadio II localizados en el recto, en los que se aconseja tratamiento combinado con quimioradioterapia adyuvante o neoadyuvante7. Por otra parte, en tumores metastásicos, en estadio IV, el tratamiento con quimioterapia también ha experimentado grandes mejoras en los últimos años, ya que las nuevas terapias combinadas permiten una importante prolongación de la supervivencia, llegando a alcanzar un tiempo medio de 16-20 meses8. Además, el tratamiento quimioterápico en pacientes en estadio IV puede realizarse en combinación con el tratamiento quirúrgico de las metástasis hepáticas o pulmonares, consiguiendo, en series de casos seleccionados supervivencias a 5 años de hasta el 40%.
PATOLOGÍA MOLECULAR DEL CÁNCER COLORRECTALLa mayoría de los CCR son esporádicos y se desarrollan a partir de la acumulación de mutaciones adquiridas en oncogenes (familia ras, c-erb B y bcl2) y genes supresores (APC, SMAD4, DCC, p53 y p27), siguiendo la vía conocida como supresora o de inestabilidad cromosómica9. En torno a un 15% de los casos de CCR esporádico se deben a una alteración epigenética en la metilación de regiones promotoras de genes reparadores de ADN. Esta alteración afecta al gen hMLH1, con la consecuente presencia del fenómeno de inestabilidad de microsatélites en estos tumores10.
Los CCR atribuibles a síndromes de cáncer hereditario suponen menos de un 5% del total de los casos11. Los síndromes mejor conocidos son la poliposis adenomatosa familiar y el CCR hereditario no asociado a poliposis (CCHNP) o síndrome de Lynch. La poliposis adenomatosa familiar se debe a la presencia de mutaciones germinales en el gen supresor APC12. El CCHNP no asociado a poliposis sigue la vía mutadora o de inestabilidad de microsatélites basada en mutaciones germinales de genes reparadores de ADN (hMLH1, hMSH2, hMSH6, hPMS1 y hPMS2)13,14. El resultado del error en la reparación del ADN es la presencia de pequeñas secuencias de ADN conocidas como microsatélites, que sirven como marcador fenotípico de estos tumores bajo el nombre de «inestabilidad de microsatélites»15.
Independientemente de la vía que haya seguido la carcinogénesis en su momento inicial, las fases posteriores de invasión y metástasis presentan una importante relación con las señales intercelulares de crecimiento celular, angiogénesis, adhesión y lisis extracelular16. Entre estas señales destacan los factores de crecimiento celular endotelial y las familias de proteasas, encargadas de la degradación de la membrana basal y la matriz extracelular.
MARCADORES PRONÓSTICO MOLECULARESEn los últimos años se han identificado múltiples marcadores moleculares relacionados tanto con el pronóstico de la evolución de la enfermedad como con la capacidad predictiva de respuesta al tratamiento quimioterápico en el CCR17,18. Este último punto es el que concierne a la revisión actual. Un buen marcador de predicción de respuesta al tratamiento quimioterápico debe identificar los tumores respondedores al fármaco estudiado, así como predecir la creación posterior de resistencias y la toxicidad durante el tratamiento. De tal forma que en la práctica clínica se plasme en una mejor respuesta al tratamiento (tiempo libre de enfermedad en tumores localmente avanzados y tiempo de progresión en tumores diseminados) y un mayor tiempo de supervivencia. Aunque hay un gran número de marcadores in vitro, no siempre se observa una buena correlación con su aplicación in vivo.
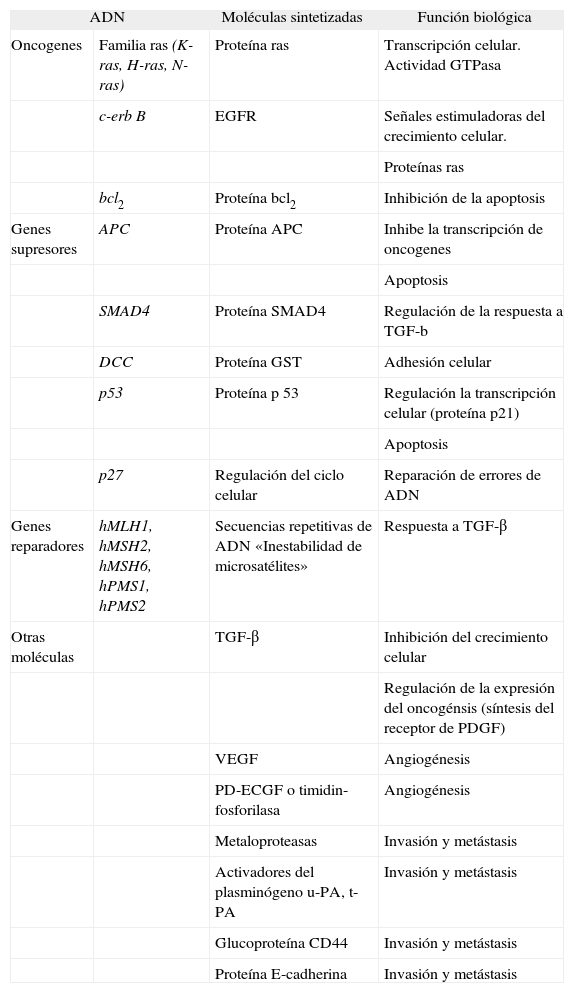
Los fármacos quimioterápicos pueden actuar sobre la carcinogénesis a distintos niveles, por lo que desde el punto de vista teórico hay múltiples marcadores de respuesta al tratamiento (tabla I). En las fases de iniciación y promoción tumoral es fundamental el estudio de las alteraciones genéticas del ADN y el ARN tumoral, así como de las proteínas tumorales sintetizadas. El análisis del ADN debe incluir la identificación de mutaciones específicas, alteraciones alélicas, amplificación, metilación, aneuploidía e inestabilidad de microsatélites. En la fase de progresión e invasión es fundamental estudiar la expresión de los factores implicados en la adhesión tisular, la angiogénesis y la destrucción de estructuras extracelulares.
Marcadores moleculares relacionados con la predicción de respuesta a tratamiento quimioterápico en el cáncer colorrectal
| ADN | Moléculas sintetizadas | Función biológica | |
| Oncogenes | Familia ras (K-ras, H-ras, N-ras) | Proteína ras | Transcripción celular. Actividad GTPasa |
| c-erb B | EGFR | Señales estimuladoras del crecimiento celular. | |
| Proteínas ras | |||
| bcl2 | Proteína bcl2 | Inhibición de la apoptosis | |
| Genes supresores | APC | Proteína APC | Inhibe la transcripción de oncogenes |
| Apoptosis | |||
| SMAD4 | Proteína SMAD4 | Regulación de la respuesta a TGF-b | |
| DCC | Proteína GST | Adhesión celular | |
| p53 | Proteína p 53 | Regulación la transcripción celular (proteína p21) | |
| Apoptosis | |||
| p27 | Regulación del ciclo celular | Reparación de errores de ADN | |
| Genes reparadores | hMLH1, hMSH2, hMSH6, hPMS1, hPMS2 | Secuencias repetitivas de ADN «Inestabilidad de microsatélites» | Respuesta a TGF-β |
| Otras moléculas | TGF-β | Inhibición del crecimiento celular | |
| Regulación de la expresión del oncogénsis (síntesis del receptor de PDGF) | |||
| VEGF | Angiogénesis | ||
| PD-ECGF o timidin-fosforilasa | Angiogénesis | ||
| Metaloproteasas | Invasión y metástasis | ||
| Activadores del plasminógeno u-PA, t-PA | Invasión y metástasis | ||
| Glucoproteína CD44 | Invasión y metástasis | ||
| Proteína E-cadherina | Invasión y metástasis | ||
EGFR: receptor del factor de crecimiento epidérmico; PD-ECGF: factor de crecimiento celular endotelial derivado de las plaquetas; PDGF: factor de crecimiento derivado de las plaquetas; t-PA: activador del plasminógeno tisular; TGF-β: factor transformante del crecimiento beta; u-PA: activador del plasminógeno de la urocinasa; VEGF: factor de crecimiento endotelial vascular.
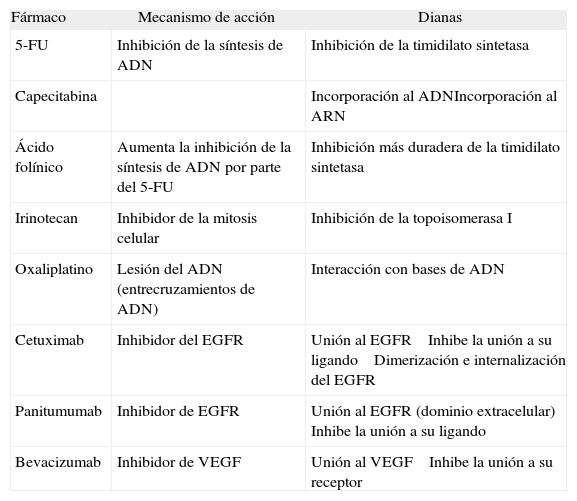
El estudio de la farmacocinética de los distintos tratamientos quimioterápicos también nos permite prever el poder predictivo de determinados marcadores moleculares implicados en el mecanismo de acción del fármaco. En la mayoría de los casos estos marcadores son las dianas sobre las que actúan el fármaco o las enzimas involu- cradas en su metabolización. En la tabla II se resume la farmacocinética de los principales fármacos quimioterápicos utilizados en el CCR.
Farmacocinética de los principales fármacos quimioterápicos utilizados en el tratamiento del cáncer colorrectal
| Fármaco | Mecanismo de acción | Dianas |
| 5-FU | Inhibición de la síntesis de ADN | Inhibición de la timidilato sintetasa |
| Capecitabina | Incorporación al ADNIncorporación al ARN | |
| Ácido folínico | Aumenta la inhibición de la síntesis de ADN por parte del 5-FU | Inhibición más duradera de la timidilato sintetasa |
| Irinotecan | Inhibidor de la mitosis celular | Inhibición de la topoisomerasa I |
| Oxaliplatino | Lesión del ADN (entrecruzamientos de ADN) | Interacción con bases de ADN |
| Cetuximab | Inhibidor del EGFR | Unión al EGFR Inhibe la unión a su ligando Dimerización e internalización del EGFR |
| Panitumumab | Inhibidor de EGFR | Unión al EGFR (dominio extracelular) Inhibe la unión a su ligando |
| Bevacizumab | Inhibidor de VEGF | Unión al VEGF Inhibe la unión a su receptor |
5-FU: 5-fluorouracilo; EGFR: receptor del factor de crecimiento epidérmico; VEGF: factor de crecimiento endotelial vascular.
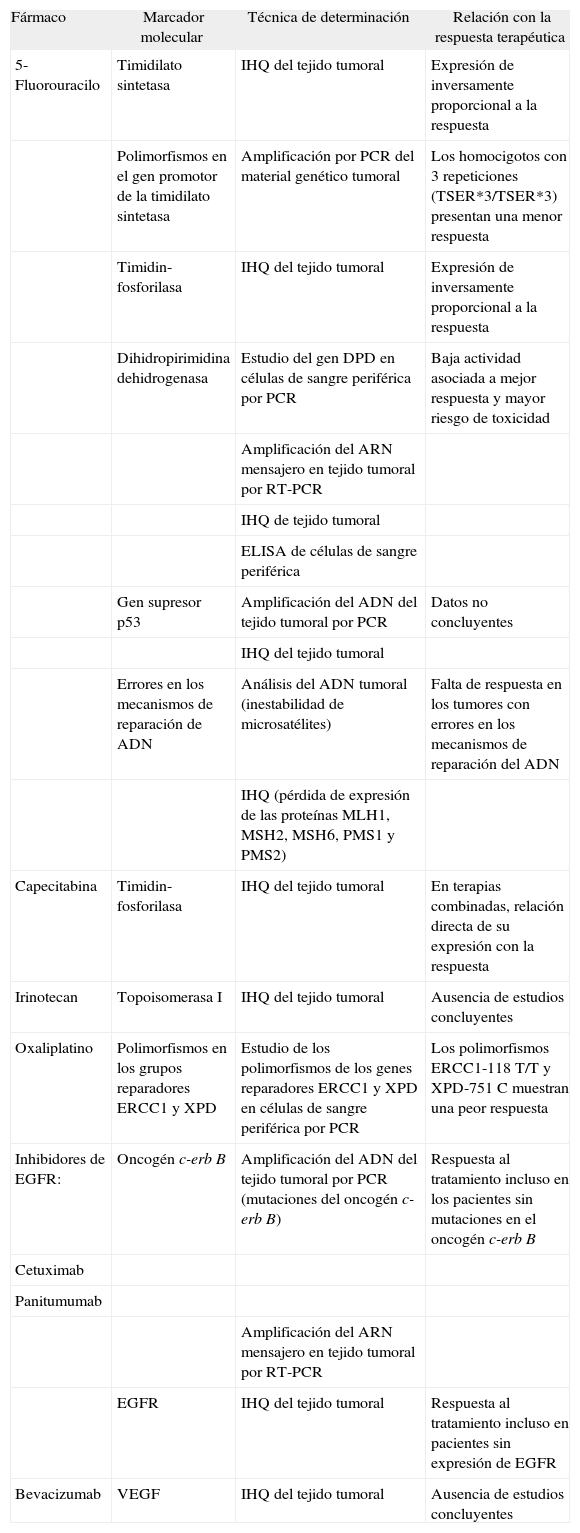
A continuación, se exponen las evidencias actuales acerca de la capacidad de predicción de los marcadores moleculares en los fármacos más utilizados en la práctica clínica, que se resumen en la tabla III.
Principales marcadores moleculares relacionados con la predicción de la respuesta al tratamiento quimioterápico en el cáncer colorrectal
| Fármaco | Marcador molecular | Técnica de determinación | Relación con la respuesta terapéutica |
| 5-Fluorouracilo | Timidilato sintetasa | IHQ del tejido tumoral | Expresión de inversamente proporcional a la respuesta |
| Polimorfismos en el gen promotor de la timidilato sintetasa | Amplificación por PCR del material genético tumoral | Los homocigotos con 3 repeticiones (TSER*3/TSER*3) presentan una menor respuesta | |
| Timidin-fosforilasa | IHQ del tejido tumoral | Expresión de inversamente proporcional a la respuesta | |
| Dihidropirimidina dehidrogenasa | Estudio del gen DPD en células de sangre periférica por PCR | Baja actividad asociada a mejor respuesta y mayor riesgo de toxicidad | |
| Amplificación del ARN mensajero en tejido tumoral por RT-PCR | |||
| IHQ de tejido tumoral | |||
| ELISA de células de sangre periférica | |||
| Gen supresor p53 | Amplificación del ADN del tejido tumoral por PCR | Datos no concluyentes | |
| IHQ del tejido tumoral | |||
| Errores en los mecanismos de reparación de ADN | Análisis del ADN tumoral (inestabilidad de microsatélites) | Falta de respuesta en los tumores con errores en los mecanismos de reparación del ADN | |
| IHQ (pérdida de expresión de las proteínas MLH1, MSH2, MSH6, PMS1 y PMS2) | |||
| Capecitabina | Timidin-fosforilasa | IHQ del tejido tumoral | En terapias combinadas, relación directa de su expresión con la respuesta |
| Irinotecan | Topoisomerasa I | IHQ del tejido tumoral | Ausencia de estudios concluyentes |
| Oxaliplatino | Polimorfismos en los grupos reparadores ERCC1 y XPD | Estudio de los polimorfismos de los genes reparadores ERCC1 y XPD en células de sangre periférica por PCR | Los polimorfismos ERCC1-118 T/T y XPD-751 C muestran una peor respuesta |
| Inhibidores de EGFR: | Oncogén c-erb B | Amplificación del ADN del tejido tumoral por PCR (mutaciones del oncogén c-erb B) | Respuesta al tratamiento incluso en los pacientes sin mutaciones en el oncogén c-erb B |
| Cetuximab | |||
| Panitumumab | |||
| Amplificación del ARN mensajero en tejido tumoral por RT-PCR | |||
| EGFR | IHQ del tejido tumoral | Respuesta al tratamiento incluso en pacientes sin expresión de EGFR | |
| Bevacizumab | VEGF | IHQ del tejido tumoral | Ausencia de estudios concluyentes |
EGFR: receptor del factor de crecimiento epidérmico; IHQ: inmunohistoquímica; PCR: reacción en cadena de la polimerasa; VEGF: factor de crecimiento endotelial vascular.
Es el grupo más importante de fármacos quimioterápicos utilizados en el CCR. Su principal exponente es el 5-FU; a pesar del desarrollo de nuevas terapias, este fármaco sigue manteniéndose como el pilar del tratamiento quimioterápico del cáncer de colon, tanto localmente avanzado como diseminado. La capecitabina es un profármaco de administración oral que ha demostrado resultados similares a 5-FU, pero con menos efectos tóxicos.
Múltiples marcadores moleculares se han relacionado con la respuesta al tratamiento con 5-FU en el CCR. Entre ellos destacan los valores de timidilato sintetasa (TS), los polimorfismos en el promotor de la timidilato sintetasa, los valores de timidin-fosforilasa (TP), los valores de dihidropirimidina deshidrogenasa, la expresión de genes supresores, como p53, y la existencia de errores en los mecanismos de reparación del ADN. A pesar de los buenos resultados de algunos de ellos, ninguno está introducido en la toma de decisiones en la práctica clínica diaria. Los valores de TS, diana principal para la acción del 5- FU, se han relacionado en múltiples estudios con la respuesta a 5-FU y con la supervivencia global en el CCR. Estos valores de TS presentan una relación inversa con la respuesta al tratamiento con 5-FU19,20, de manera que a mayor valor de TS, menor será la respuesta al tratamiento con 5-FU. Una aproximación semicuantitativa de los valores de TS se puede realizar de manera sencilla mediante técnicas de inmunohistoquímica sobre el tejido tumoral21.
Los polimorfismos en el gen promotor de la TS, determinados por amplificación por reacción en cadena de la polimerasa (PCR) del ADN tumoral, guardan relación con los valores de TS. Los pacientes homocigotos con 3 repeticiones (TSER*3/TSER*3) presentan una mayor actividad TS y, por tanto, una menor respuesta al tratamiento con 5-FU que los homocigotos con doble repetición (TSER*2/TSER*2) o los heterocigotos (TSER*3/ TSER*2)22,23.
La TP, o también llamada factor de crecimiento celular endotelial derivado de las plaquetas (PD-EGF), es la enzima que cataliza la fosforilización intracelular de la timidina a 2-deoxy-D-ribosa, y contribuye a la conversión de la capecitabina en 5-FU24. Además de contribuir a la activación de la capecitabina, la TP participa en la angiogénesis tumoral precoz. Este papel angiogénico podría ser responsable de la relación inversa de la expresión genética de TP y la respuesta al tratamiento con 5-FU25. Respecto a la capecitabina, aunque hay estudios con terapias combinadas que muestran una relación directa entre los valores de TP y la respuesta al tratamiento, se carece de estudios en monoterapia26. El reciente desarrollo de técnicas de inmunohistoquímica sencillas, junto con la necesidad de definir mejor el papel de esta enzima como marcador predictivo, hace necesarios nuevos estudios en este campo27.
La dihidropirimidina deshidrogenasa (DPD) es una enzima muy importante en la vía catabólica del 5-FU, cuya actividad presenta importantes variaciones individuales28,29. La baja actividad de esta enzima se asocia a mejores respuestas al tratamiento con 5-FU, aunque con un mayor riesgo de toxicidad30,31. Esto se explica porque la disminución en la inactivación del 5-FU dirige su metabolismo hacia la vía anabólica con creación de metabolitos activos citotóxicos32. Por las graves consecuencias que podría tener sobre la toxicidad por 5-FU, la determinación de esta enzima tendría relevancia clínica en la identificación de la población con alto riesgo de toxicidad por 5-FU. La actividad de DPD se puede determinar directamente en el tejido tumoral y en las células de la sangre periférica o, de forma indirecta, mediante el estudio del gen DPD en sangre periférica o de su ARN mensajero en tejido tumoral33,34.
El gen supresor p53 presenta mutaciones en un 50% de los CCR, que conducen a una expresión de la proteína p53 anómala. Ésta presenta una vida media más duradera, por lo que en la inmunohistoquímica se detecta como una sobreexpresión de p53. La relación de la presencia de mutaciones en el gen p53 y la respuesta al tratamiento quimioterápico permanece en discusión. Mientras algunos estudios apoyan la relación de una sobreexpresión anómala de p53 con una peor respuesta al tratamiento quimioterápico35,36, otros niegan esta relación37,38. El estudio de las mutaciones del gen supresor p53 se puede realizar mediante la amplificación de ADN del tejido tumoral o mediante evaluación de la sobreexpresión de la proteína p53 anómala en tejido tumoral por inmunohistoquímica.
Los errores en los mecanismos de reparación del ADN, ya sean congénitos o adquiridos, se asocian a una falta de respuesta al tratamiento con 5-FU. Diversos estudios retrospectivos39–41 y un estudio prospectivo anidado en el proyecto EPICOLON42,43 han demostrado que los tumores en estadios II o III, con alteraciones en el sistema reparador de errores en el ADN, no mejoran su pronóstico tras ser tratados con 5-FU. La consistencia aportada por estos estudios es suficiente para recomendar, al menos, la valoración de otros tratamientos quimioterápicos adyuvantes en los pacientes con errores en los mecanismos de reparación44. Esto sucede porque el sistema reparador de errores en el ADN es necesario para el correcto reconocimiento del 5-FU y su incorporación al ADN. Si este sistema no funciona, el 5-FU no se incorpora al ADN de las células tumorales y no ejerce su efecto citotóxico sobre estas células. Los errores en los mecanismos de reparación del ADN afectan a la práctica totalidad de los tumores del síndrome de Lynch y a un 15% de los CCR esporádicos, por lo que este marcador puede ser de gran importancia a la hora de tomar decisiones acerca de tratamiento con 5-FU de estos pacientes. Además, hay notables evidencias que indican que la presencia de alteración en el sistema reparador de errores en el ADN es un marcador de buen pronóstico en pacientes con CCR45. De este modo, la suma de indicador de buen pronóstico y factor predictivo de poca eficacia de tratamiento con 5-FU hacen que este marcador pueda ser especialmente interesante. En este sentido, es probable que los pacientes en estadio II, con alteración en el sistema reparador de errores en el ADN, deban considerarse de bajo riesgo y, por tanto, no tratarlos con quimioterapia adyuvante. Por otra parte, los pacientes en estadio III deberían probablemente ser tratados con otros fármacos diferentes del 5-FU. En cualquier caso, ensayos clínicos que se están desarrollando podrán definir finalmente el papel de este marcador en la toma de decisiones acerca del tratamiento en pacientes con CCR. La alteración en el sistema reparador de errores en el ADN puede ponerse de manifiesto mediante técnicas sencillas que abarcan desde el análisis del ADN tumoral en busca del fenómeno de inestabilidad de microsatélites hasta el estudio inmunohistoquímico del tejido tumoral, con el fin de detectar la pérdida de expresión de alguna de las proteínas reparado- ras del ADN, como MLH1, MSH2, MSH6, PMS1 y PMS2.
IrinotecanEl irinotecan es un fármaco inhibidor de la topoisomerasa I utilizado en el tratamiento del cáncer de colon metastático. La topoisomerasa I puede ser valorada de forma muy sencilla mediante técnicas de inmunohistoquímica en el tejido tumoral45. Aunque la expresión topoisomerasa I en el tumor podría tener relación con su repuesta al tratamiento con irinotecan, se carece de estudios que confirmen esta relación. Por otra parte, los estudios realizados en xenoinjertos y en humanos han mostrado la posibilidad de un mayor efecto del irinotecan en tumores con alteración en el sistema reparador de errores en el ADN47.
OxaliplatinoEl oxaliplatino es un fármaco que, utilizado en asociación con el 5-FU, constituye el estándar actual de tratamiento del CCR localmente avanzado y diseminado. El oxaliplatino produce lesiones por entrecruzamiento en el ADN tumoral. Estas lesiones se pueden subsanar mediante la vía reparadora por escisión de nucleótidos gracias a los grupos reparadores ERCC1 y XPD48. Determinados polimorfismos en estos grupos reparadores se han relacionado con una menor respuesta al tratamiento combinado con oxaliplatino y 5-FU49. La determinación de polimorfismos se realiza mediante la PCR del ADN extraído de células de sangre periférica. Algunos estudios in vitro han mostrado una asociación entre los defectos en el sistema reparador de errores en el ADN y la resistencia a derivados de platino, probablemente relacionada con la necesidad de este sistema para el correcto funcionamiento de los compuestos de platino50.
Inhibidores del receptor del factor del crecimiento epidérmico (EGFR): cetuximab y panitumumabLos inhibidores del EGFR son anticuerpos monoclonales utilizados en el cáncer de colon diseminado. El EGFR es un receptor tirosin-cinasa codificado por el oncogén c-erb B, implicado en las señales estimuladoras del crecimiento celular. Un 80% de los CCR muestran sobreexpresión de EGFR. El estudio de la expresión de EGFR se puede realizar mediante la secuenciación de las mutaciones del oncogén c-erb B, el estudio del ARN mensajero o la inmunohistoquímica del EGFR en el tejido tumoral. Aunque la indicación inicial de este tratamiento requería constatar la sobreexpresión del receptor en tejido tumoral, algunos estudios recientes han mostrado respuestas en tumores sin expresión de EGFR y tumores sin mutaciones en el oncogén c-erb B50. Por esta razón, actualmente no se requiere valorar la expresión de EGFR previa al tratamiento con sus inhibidores y, por tanto, no se puede considerar éste como un marcador predictor de respuesta. Sin embargo, recientemente se ha sugerido la utilidad del estudio de la existencia de mutación KRAS en tejido tumoral como predictor de falta de respuesta a cetuximab y panitumumab en el CCR avanzado. De esta manera los tumores que mostraban la forma salvaje del gen KRAS tenían mucha mejor respuesta a los inhibidores de EGFR que los que tenían el gen KRAS mutado52,53.
BevacizumabEl bevacizumab es un inhibidor del factor de crecimiento endotelial vascular (VEGF), utilizado junto con 5-FU, oxaliplatino o irinotecan en el tratamiento del cáncer de colon avanzado. El VEGF es un factor de crecimiento crucial para el desarrollo de la angiogénesis. Su sobreexpresión está presente en aproximadamente la mitad de los cánceres de colon. La inmunohistoquímica es la técnica de elección para valorar esta sobreexpresión. Actualmente, se carece de estudios sobre la validez de la expresión del VEGF en la respuesta al tratamiento quimioterápico en el cáncer de colon.
CONCLUSIONESLa heterogeneidad de los estudios impide la estandarización del uso de los marcadores moleculares como pilar en la elección del tratamiento quimioterápico. Por tanto, se requieren nuevos estudios prospectivos que comparen la respuesta del tratamiento quimioterápico en relación con el uso o no de los marcadores moleculares, realizando un subanálisis de acuerdo a los distintos estadios tumorales. A pesar de estas limitaciones, el papel de los marcadores moleculares en la toma de decisiones acerca del tratamiento del CCR será, sin duda, de importancia creciente en los próximos años.
recomendados


Gastroenterología y Hepatología sigue las recomendaciones para la preparación, presentación y publicación de trabajos académicos en revistas biomédicas