







Introduction
In 1962, during the process of synthesis and purification of chloroquine (an anti-malaria agent), a quinolone derivative, nalidixic acid, was discovered which was active against gram-negative bacteria. This agent was able to reach high concentrations in urine1. However, it was only used in the treatment of urinary tract infections (URTI). The addition of a fluor atom in position 6 of the quinolone molecule increased its activity, but it was not until the end of the 1980s and the beginning of the 1990s when new fluoroquinolones with activity against gram-negative and gram-positive bacteria, including anaerobes, were introduced in clinics2.
At present, fluoroquinolones are used in various types of infections, including bacteremias, respiratory tract infections, osteomyelitis, enteral and gonococal infections,3 but they also have prophylactic use, for example with neutrophenic patients (although the risk of developing resistance to gram-negative bacilli during this type of program is high). Quinolones, together with other antibacterial agents, have also been used in the veterinary environment.
The action mechanism in fluoroquinolones is quite complex. This kind of agent penetrates gram-negative bacteria through the porins although it is also capable of direct activity through the lipid membrane, then crossing the internal membrane in order to reach the cytoplasm. In gram-positive bacteria, penetration occurs directly through its wrapped cell until it reaches the cytoplasm. Subsequently, it acts at the level of the bacterial DNA by inhibiting the topoisomerases (DNA gyrase and topoisomerase IV). When the fluoroquinolone unites with the subunits of the DNA gyrase, loose DNA ends appear on which exonucleases act, thus bringing about cell death4,5. This last mechanism of antibacterial action is not well understood.
Extensive use of antimicrobial agents has generated the appearance of bacteria resistant to them. To date, the main mechanisms implied in this resistance have been two (both by elements chromosomal): alterations in the targets of the quinolones and decrease in the accumulation of the antibiotic in the bacterial interior by making the membrane impermeable (loss of porins or alterations of the lipopolysaccharide) or by expression of active expulsion systems. In 1998, horizontally transferable resistance to quinolones was described for the first time6. The qnr gene is genetically responsible for resistance and is found inside a mobile element. The horizontal dissemination of mechanisms resistant to fluoroquinolones opens up the possibility of a rapid expansion of resistance to such antimicrobials, in animal as well as human pathogens, and even more so with the extensive use made of quinolones.
Mechanisms of action and chromosomal resistance.
Fluoroquinolones inhibit DNA synthesis when the antibiotic interacts with the complex formed by the union of the DNA with the quinolone target, the DNA gyrase and topoisomerase IV. These two enzymes are structurally related, being composed of two different pairs of subunits: GyrA and GyrB (in the case of DNA gyrase) and ParC and ParE (in the case of topoisomerase IV). Both enzymes are type II topoisomerases, which act by cutting both chains of a DNA segment, passing another DNA segment through the break and returning to unite the loose ends (Figure 1). DNA gyrase is implicated in the loosening or coiling of the DNA, for example, during DNA synthesis. Topoisomerase IV is implicated in the separation of the daughter chromosomes after DNA replication. In both cases, the fluoroquinolone acts by trapping enzymes in the DNA during the topoisomerization reaction, after the enzyme has broken the DNA and generated loose ends. Union with the antimicrobial stabilizes the complex, creating a physical barrier to movement of the replication fork8 the RNA polymerase9 and the DNA helicase10. The collision of this complex with the replication fork unleashes a series of events, including the activation of the SOS system (which is not, in general, well understood) and whose final result is cell death.
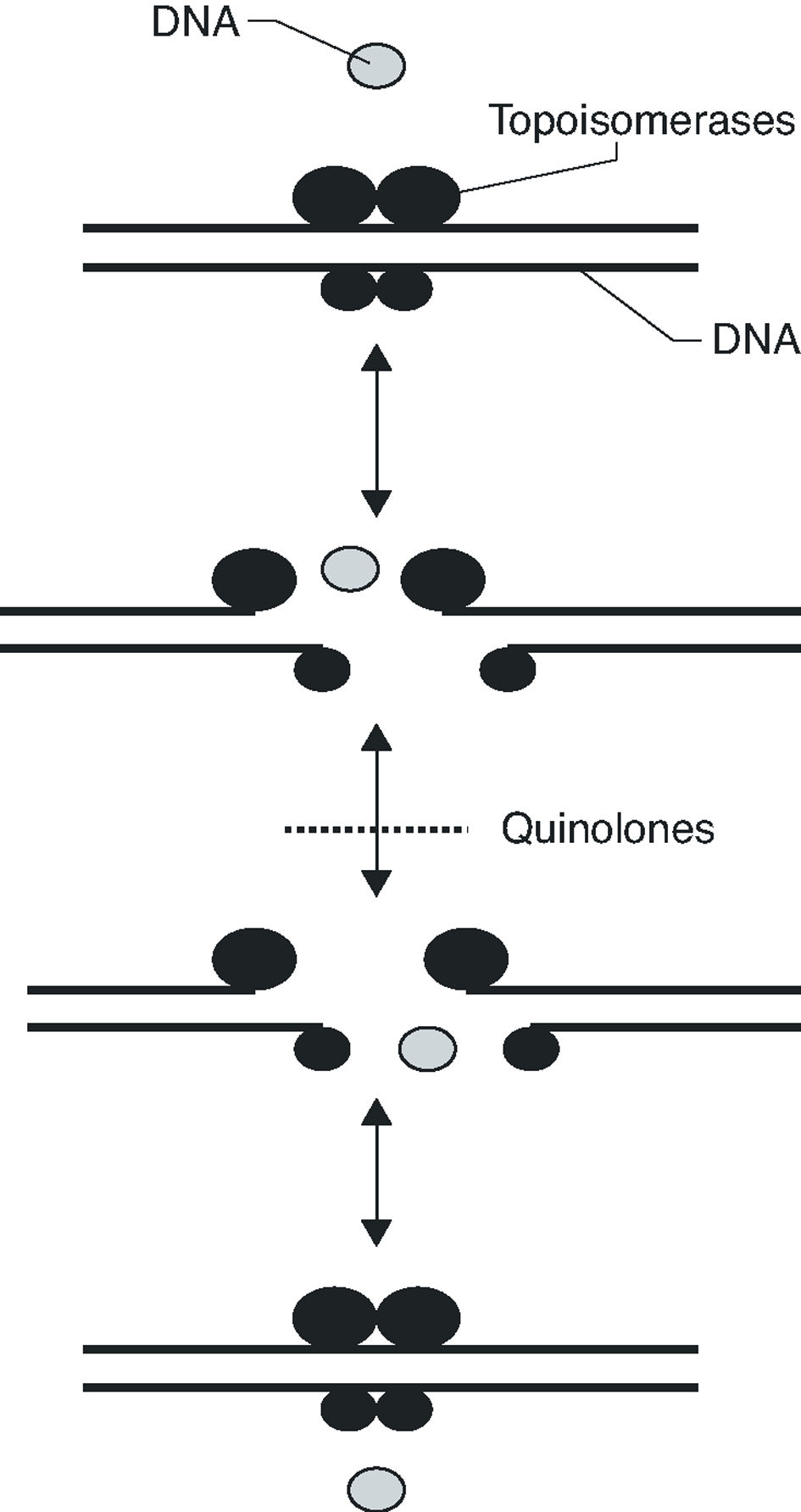
Figure 1. Activity mechanism of type II topoisomerases.
The quinolone target depends on the drug under consideration and may vary according to whether it is gram-positive or gram-negative. If it is gram-positive, the primary target is, in many cases, topoisomerase IV; if gram-negative, it is the DNA gyrase. More recently developed fluoroquinolones, such as moxifloxacin or clinafloxacin, present a similar affinity for both targets.11
To summarise, the quinolone acts in four phases: it passes through the porins in the bacterial wall; crosses the membrane; inhibits the DNA gyrase and/or topoisomerase IV; and induces the SOS system (Figure 2).
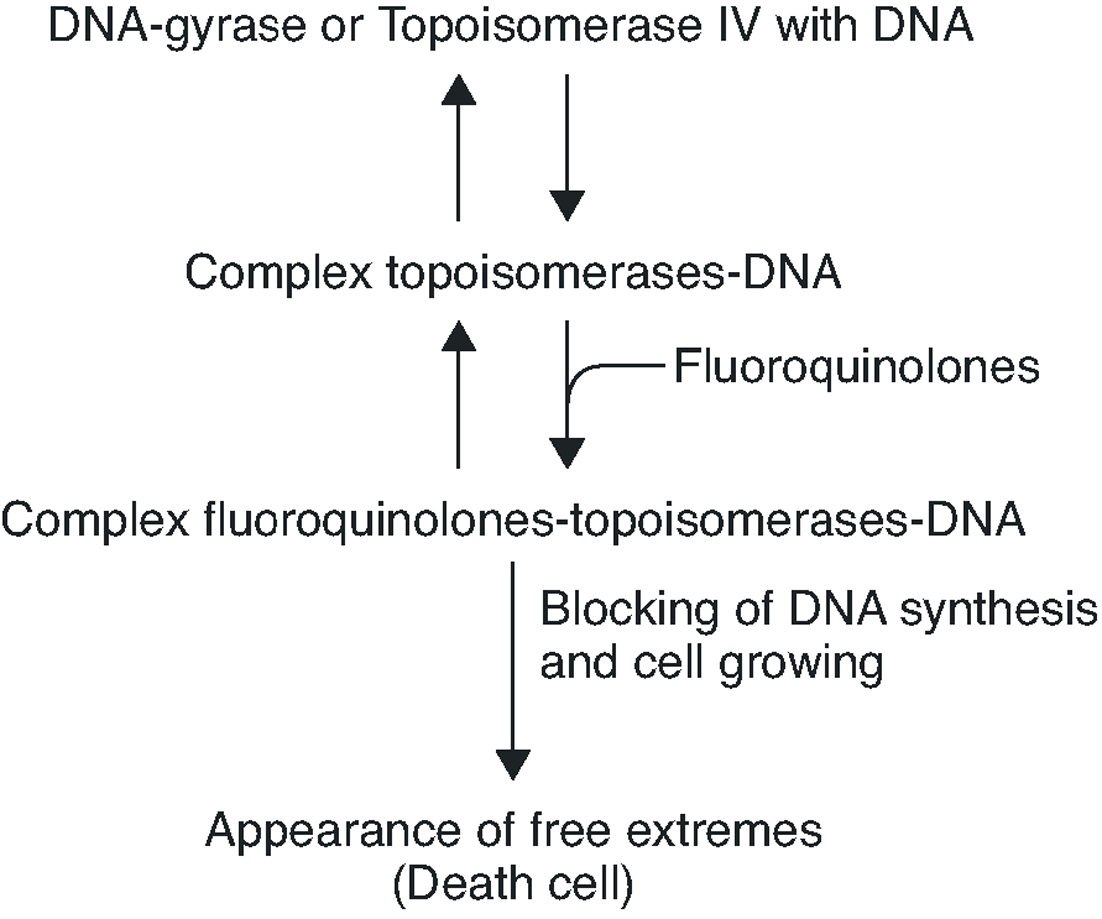
Figure 2. Interruption of topoisomerase activity by quinolones.
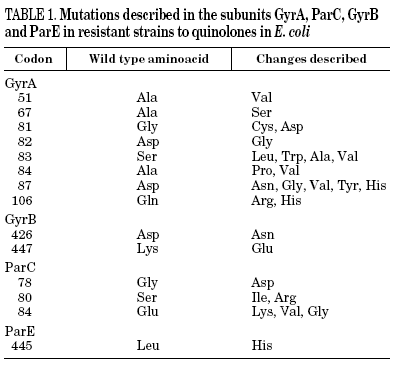
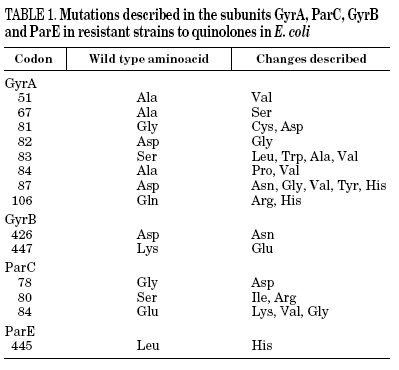
Resistance to quinolones in gram-negative bacteria is mainly caused by chromosomal mutations4,5,7. In E. coli, target modification is determined by gyrA or parC mutations (which code for the A subunit of DNA gyrase and topoisomerase IV, respectively), and, to a lesser extent, by gyrB or parE (which code for the B subunit of DNA gyrase and topoisomerase IV, respectively). Crystallographic studies of the GyrA structure suggest that aminoacid changes occur in the active site of the region where the enzyme unites with the DNA and interacts with the quinolone12 (Table 1). It has been shown, for both E. coli and S. aureus, that different levels of quinolone resistance depend on whether the alterations take place in the primary target, the secondary one, or both.
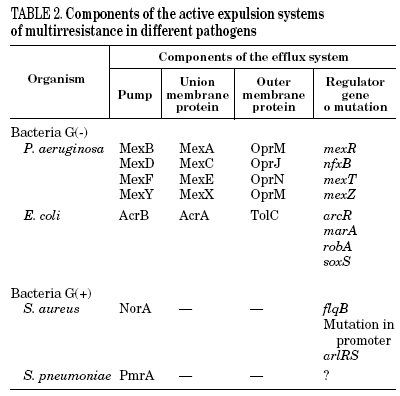
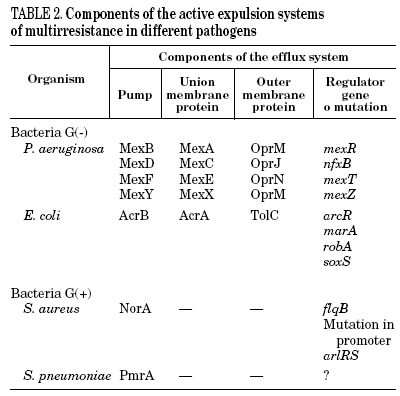
Mutations in the active expulsion pumps responsible for eliminating toxic compounds (due to expression or overexpression causing simultaneous resistance to different groups of antibiotics) are another important cause of fluoroquinolone resistance. These systems are in many, but not all bacteria. In some microorganisms, such as Stenotrophomonas maltophilia, this mechanism may be of great importance.13 The norA gene codes for a pump that contributes to resistance in S. aureus,14 whilst acrAB in E. coli codes for a multidrug expulsion system, that associated to product of tolC, modulates resistance to quinolones in this species.15 Overexpression of NorA by a mutation in the promoter causes a two to fourfold increase in the MIC of ciprofloxacin.16 It seems that the expression of simple active expulsion pumps has a limited effect on fluoroquinolone resistance, but its base expression contributes notably to resistance caused by other mechanisms. For example, in strains with mutations in gyrA and acrB, the level of resistance diminishes considerably. On the other hand, a large number of pumps, potentially implicated in resistance, have been characterized, so that it is difficult to define the role of each one of them with any accuracy (Table 2).
Loss of membrane permeability is another form of resistance, brought about by mutations in the structural or regulating genes which reduce the effective number of porins (ompC and ompF genes in E. coli; ompK35 and ompK36 genes in K. pneumoniae).
Recently, low levels of resistance caused by the reduced expression of topoisomerase IV in S. aureus have been reported for the first time. Reduced levels of ParE are compatible with bacterial survival, although this ought to imply a cost in the speed of cell division. It seems that this phenomenon should entail other associated compensating mechanisms which do not revert the phenotype of resistance.17
Potential mechanisms of plasmid-mediated quinolone resistance
In principle, the hereditary nature of the chromosomal mutations that cause quinolone resistance would mean that such mutants do not need horizontally transferable resistance mechanisms, since their survival and vertical dissemination are ensured. In other words, since the receptor strain is quinolone resistant, it ought not to require any other gene of resistance.
Plasmids with chromosomal genes
One possibility that has been speculatively put forward is the acquisition of the chromosomal genes of DNA gyrase or topoisomerase IV with quinolone resistant mutations by means of plasmids, or mobile elements. Furthermore, high rates of resistance in gram-positive bacteria, evidence of the natural transfer of genetic material from gram-positive to gram-negative18 and the absence of barriers to the expression of gram-positive genes in gram-negative ones19 open up a theoretical means of transferable quinolone resistance. However, in many species, the presence of a mutant topoisomerase is recessive against the wild topoisomerase of the microorganism.
Inactivation of the drug
Inactivation of the drug (by oxidisation, reduction, sterification or other reactions) is the most common resistance mechanism amongst bacterial pathogens, producing cross resistance to different antimicrobials of the same group. Up to now, only fungi have been reported as capable of degrading quinolones.20,21. Since these compounds are synthetic products of the laboratory and not themselves produced by bacteria or mushrooms, it is difficult to believe that there exists a natural process which exerts pressure against quinolones in environmental or pathogenic microorganisms. However, quinolones may be inactivated by enzymes implicated in the degradation of other more or less related compounds, and for which environmental pressure exists.
Mechanisms of qnr action
In 1987, plasmid-mediated quinolone resistance was reported in a Shigella dysenteriae strain,22 which could not subsequently be verified. In 1998 came the first published report of the existence of a clinical strain of K. pneumoniae, isolated in a urine culture collected in Birmingham, Alabama (USA), containing a plasmid with a wide range of hosts, and whose transconjugants, in E. coli, increased resistance to nalixidic acid from 4 to 32 mg/L and to ciprofloxacin from 0.008 to 0.25 mg/L23. This plasmid, named pMG252, increased resistance between 4 and 8 times due to particular mutations in the DNA-gyrase, porins or active expulsion pumps,24 and facilitated the selection of mutants resistant to quinolones, by mechanisms which are not yet known. The presence of this plasmid did not alter the pattern of porin expression in the host neither did it reduce the accumulation of quinolones, which suggested the existence of a new resistance mechanism.
To clarify this possible new mechanism, the qnr gene of pMG252 was cloned, sequenced, later amplified by PCR and introduced into a vector of expression. In this way, the protein encoded by qnr was purified and the interaction between quinolone and target studied by electrophoretic mobility assay. It enabled researchers to show that, at least in vitro, qnr protects the DNA-gyrase of E. coli from inhibition by ciprofloxacin25. This protection is proportional to the concentration of qnr and inversely proportional to the concentration of ciprofloxacin.25 Topoisomerase IV, the secondary target of the quinolone in E. coli, also seems to be protected from quinolones by qnr26.
Qnr belongs to the family of pentapeptide repeats, of which more than 90 members are known. This family is defined by the presence of repetitions in tandem of the pattern A(D/N)LXX, where X is any aminoacid27. These proteins have been found in many bacteria, but seem particularly common in cyanobacteria, where they are able to be membrane proteins as well as cytoplasmic ones. These proteins present a *-helix structure in their external circumference and of ß parallel leaves in their internal circumference,28 an appropriate structure for interaction between proteins.
In the pentapeptide family, there are two members of special importance in quinolone resistance. The first is McbG, a protein that protects bacteria which synthesize microcin B17 (MccB17) from self-inhibition. MccB17 is a postascriptionally modified peptide of 3.1 kDa that blocks DNA replication,29 and is able, like ciprofloxacin, to inhibit the activity of the DNA-gyrase,30 to stabilize the DNA-DNA-gyrase complex in the presence of ATP31 and of loose ends of DNA. The self-immunity mechanism conferred by mcbG involves other genes, mcbE and mcbF, related to the expulsion of MccB17 from the cell.32 It has been verified that a plasmid carrying the mcbEFG operon produces a two to eightfold increase in the MIC of quinolones.33 In relation to this system, a new protein, SbmC, was reported in 2002 which also protects E. coli from the activity of MccB17.34
In E. coli, the mcb operon (responsible for producing MccB17) and the emr operon (which codes for the EmrAB resistance pump to multiple compounds) share the same repressor: EmrR. In addition, the compounds that induce the operon emr repress the operon mcb.33
The second member of the pentapeptide family is MfpA, a protein cloned from the genome of Mycobacterium smegmatis in studies of active expulsion pumps that contribute to quinolone resistance.35 The artificial plasmids that code for MfpA increase resistance to ciprofloxacin 4 times. The resistance mechanism has still not been established, but it is known that MfpA has no effect on the accumulation of ciprofloxacin marked with C14.
The relation between the members of this family and qnr is difficult to establish, amongst other reasons, because the percentage of homology between qnr and McbG or MfpA is 19.6% and 18.9%, respectively25. Using existing data, we can only speculate that qnr has appeared from some protein of immunity designed to protect the DNA-gyrase from natural inhibitors, or from some chromosomal gene of unknown function that codes for a protein of the pentapeptide family in the mycobacterial, cyanobacterial or other bacterial group.
Genetic environment of qnr
The qnr gene is found, in the strains in which it has been reported, in plasmids transferable by conjugation. Studies of plasmid pMG252, carried out when it was first described, reveal that qnr is found in a plasmid with an extensive range of hosts, transferable by conjugation in species such as K. pneumoniae, E. coli, C. freundii, S. typhimurium and P. aeruginos36. In the original plasmid in which it was isolated, qnr was located as forming part of a nucleotide sequence originally characteristic of integrons In6 (of the pSa plasmid) and In7 (of pDGO100),37 and suggesting its presence in a class 1 integron.25 These integrons possess a common 3'-conserved region containing the qacE1 gene (which confers a low level of resistance to certain amonic compounds)38 and the sulI gene (which confers a low level of resistance to sulfonamides, but which does not express itself in integrons due to the loss of its promoter).39 An unusual number of class 1 integrons that contain the common region of In6 and In7 carry an element, initially called orf341 and now orf513, which, it is postulated, codes for a site-specific recombinase for the acquisition of gene resistance.39 Many genes of resistance, such as the one which codes for beta-lactamase plasmids, are found located within such mobile elements, and also inside transposons, which, as is well known, increase dissemination.40 In these resistance cassettes, including qnr, the element of 59 bp41 has been lost, indicating that orf513 should really be implicated in the site-specific acquisition of genes.
It is important to indicate that there exists a statistically significant relation between quinolone resistance and beta-lactams,42 and pMG252 contains the extended spectrum beta-lactamase, FOX-5 25. Thus, a means of co-resistance is opened up to two very important families of antimicrobials, particularly in strains with some mechanism of quinolone resistance, such as loss of porins, in which the presence of qnr enables high levels of resistance to be reached.
In a more recent study,43 an analysis was carried out of the genetic environment of the transconjugants of two quinolone-resistant strains of E. coli, in a hospital in Shanghai in which qnr had been identified. In these strains, qnr forms part of a class 1 integron, belonging to the In4 family, adjacent to orf513 and upstream of ampR, qacED1 and sul1 (Figure 3). The qnr gene of the original pMG252 plasmid has a similar location, although in the plasmids from the Shanghai strains, the ampR gene is found immediately downstream of qnr while in pMG252, qacED1 and sul1 are directly downstream of qnr. In the two new plasmids, the integrons that contain qnr, In36 and In37,43 presented a similar structure to that of pSal-144, but ampC is substituted by qnr (Figure 3). In the remaining qnr positive strains from the study of the Shanghai strains, PCR verified that ampR is found next to orf513, but ampC has been substituted by qnr.
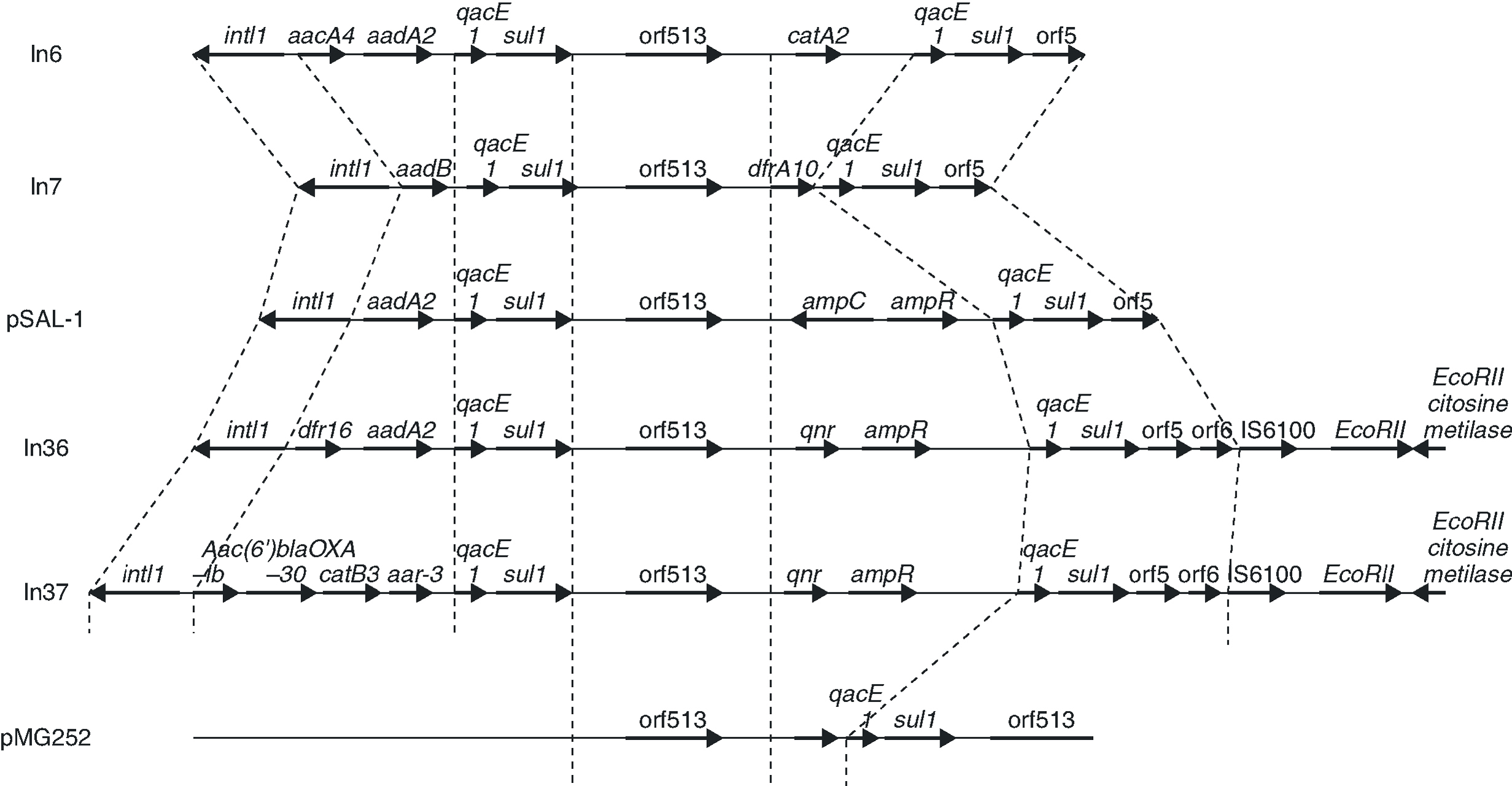
Figure 3. The genetic environment of strains in which qnr has been found and sequence comparison with other class 1 integrons.
Integron mobility is frequently due to its presence in transposons or plasmids. The integrons that carry qnr seem to be mobile between different plasmids. This interpretation is supported by several observations. First, in the clinical strains in which it has been detected, qnr is found in plasmids of different sizes; second, one of the strains is found in two plasmids of different sizes (in addition, these plasmids are able to transfer qnr by conjugation).43 Third, in some transconjugants, the hybridization of qnr is lost. Without the presence of the plasmid, it is lost in high temperatures or in the absence of antibiotic pressure 43. The mechanism which mobilises integrons is not clear, but their mobility is supported by multiple localizations which indicate past movement, and by the presence, in some of them, of direct repeats of 5 bp, consistent with movement by some mechanism of transposition.45 It seems reasonable to think that class 1 integrons can be mobilized if they possess the characteristic sequences of IRi and IRt, and that the tni genes, which code for transposition enzymes, are contributed in trans from other structures, plasmid or chromosomal45. The two integrons reported as containing qnr, In36 and In37, also contain both IRi and IRt sequences as direct duplications of 5 bp, indicating that movement by transposition is possible.43 The mobility of qnr could be facilitated in these clinical strains by the presence of genes tni in another transposon.
Effect of Qnr on the activity of quinolones.
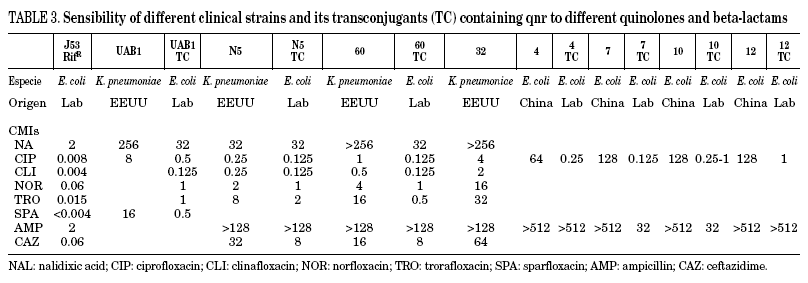
In Table 3, the MICs of different quinolones can be seen against some of the clinical strains published and transconjugants obtained. As can be seen, the MICs of ciprofloxacin against the transconjugants are between 0.125 and 2 mg/L, representing an increase in MIC of 16 to 250 times when compared to the donors. In a recent study,46 the activities of different quinolones were tested against qnr, and it was seen that antimicrobials such as sitafloxacin, BAYy3118 and premafloxacin are more active than ciprofloxacin, in both transconjugant as well as donor.
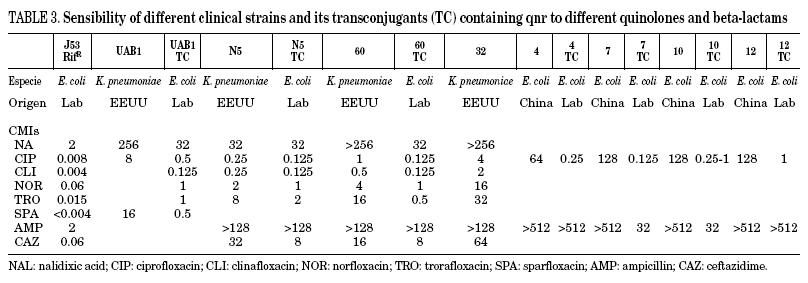
The presence of qnr in the transconjugants analysed was stable after successive growth in media without antibiotic pressure, retaining the phenotype of resistance to quinolones, accompanied by other resistances, except in a case where quinolone resistance is lost, and also the associated to trimethoprim+sulfamethoxazol, chloramphenicol and tetracycline.43 The particular plasmid that possessed this transconjugant was of a slightly smaller size than expected, indicating that, at least in this case, part of the integron might have been lost during the conjugation process.43
Although qnr produces low levels of quinolone resistance, it also facilitates selection for a high level of quinolone resistance.47 It has been known for some time that the plasmids that mediate resistance to low concentrations of streptomycin increase selection for greater resistance.48 Thus, when qnr is expressed in porin-deficient strains, the MICs against ciprofloxacin, levofloxacin and moxifloxacin increase from 8 to 32 times,49 reaching MICs of between 0.25-0.5 and 4-8 mg/L. An additional effect of the different mechanisms of quinolone resistance exists with the presence of qnr.50 It is thought that low resistance to antimicrobials enables the bacterial population to reach concentrations where secondary mutations appear, enabling high resistance. This could be what took place with resistance to both streptomycin and quinolones encoded by pMG252, the original plasmid in which qnr was reported, and where the frequency of a high resistance mutation increases in transconjugants with respect to the E. coli J53 host without a plasmid47.
Prevalence of qnr
Integrons are common elements in clinical isolates in gram-negative bacteria, being present in more than 40% of them. Furthermore, these isolates are significantly more resistant to antibiotics such as quinolones, aminoglycosides and beta-lactams51, suggesting the possibility that qnr, or another related gene present there, may be found in strains of this type, many with the conserved sequences of class 1 integrons.
Not enough studies have been carried out to determine the actual prevalence of the horizontally transferable qnr determinant of quinolone resistance. In an early study,52 out of a total of 350 strains of 13 different genera of gram-negative bacteria, mostly originating in the US (only one of which was E. coli), four isolates of K. pneumoniae and one isolate of Klebsiella sp presented the qnr gene. In a second study,43 out of 78 clinical strains of E. coli resistant to quinolones, collected in 5 hospitals in Shanghai, China, from March 2000 to March 2001, 7.7% (6 strains) possessed the qnr gene. In this case, the strains proceeded from different patients of the same hospital. In a third study,53 of a total of 266 strains of E. coli and 159 strains of K. pnuemoniae with different phenotypes of resistance to beta-lactams and quinolones, the results showed that three strains of K. pneumoniae contained the qnr gene, whilst none of the E. coli strains was positive. Recently, Wang et al54 have published a work on clinical strains of K. pneumoniae and E. coli from different states in the US, in which the frequency of qnr is greater than 11% in K. pneumoniae (qnr was not detected in any strain of E. coli). Some of these strains produced SHV-7, this being the first report of an association between an extended spectrum beta-lactamase and qnr for the same strain. From these studies, it can be deduced that this gene is extensively distributed in quinolone resistant clinical strains of K. pneumoniae in the US and of E. coli in southeast Asia.
Conclusion and future perspectives
The identification of qnr in clinical strains of K. pneumoniae isolated in the US and producing beta-lactamase plasmids, and its discovery in strains of E. coli originating from southeast Asia indicate the emergence of this new mechanism of quinolone resistance in clinical strains. Furthermore, the possibility of its dissemination to other genera of gram-negative bacteria would aggravate the problem. qnr exerts quinolone resistance from class 1 integrons, united with other genes of resistance which has an additional effect, since the reduction of sensibility to quinolones in the presence of other resistance mechanisms promotes selection of genes of this type found in the same integron. Thus, new studies are necessary to clarify the actual prevalence of this mechanism (including gram-positive ones), mechanisms of resistance and mutant selection in the presence of qnr, as well as test animals to study its clinical significance in vivo.

