









Introducción
En 1962, durante el proceso de síntesis y purificación de la cloroquina (un agente contra la malaria) se descubrió un derivado de las quinolonas, el ácido nalidíxico, activo frente a algunas bacterias gramnegativas y capaz de alcanzar elevadas concentraciones en orina1. Sin embargo, su uso clínico se limitó al tratamiento de infecciones del tracto urinario (ITU). La adición de un átomo de flúor en la posición 6 de las moléculas de quinolonas potenció en gran medida su actividad, pero no fue hasta fines de la década de 1980 y principios de 1990 cuando se introdujeron en clínica nuevas fluoroquinolonas con actividad frente a bacterias gramnegativas y a grampositivas, incluso frente a anaerobias2.
En nuestros días, las fluoroquinolonas se usan en diversos tipos de infecciones, que incluyen bacteriemias, infecciones del tracto respiratorio, osteomielitis, infecciones entéricas o infecciones gonocócicas3 y, por otro lado, tienen un uso profiláctico por ejemplo en pacientes neutropénicos (aunque el riesgo de desarrollo de resistencia en bacilos gramnegativos durante este tipo de programas es alta). Además, las quinolonas, junto con otros agentes antibacterianos, se han usado en el ámbito veterinario.
El mecanismo de acción de las fluoroquinolonas es bastante complejo. Este tipo de fármacos penetran en bacterias gramnegativas a través de porinas, pudiendo también hacerlo directamente a través de la bicapa lipídica, y luego atraviesan la membrana interna para alcanzar el citoplasma. En bacterias grampositivas la penetración ocurre directamente a través de la envuelta celular hasta alcanzar el citoplasma. Posteriormente actúan a nivel del ADN bacteriano produciendo la inhibición de las topoisomerasas (ADN-girasa y topoisomerasa IV). Al unirse las fluoroquinolonas a las subunidades de la ADN-girasa se produce la aparición de extremos libres de ADN, sobre los cuales actuarán exonucleasas que producirán la muerte celular4,5. El mecanismo último de acción bactericida no se conoce bien.
El extenso uso de estos antimicrobianos ha generado la aparición de bacterias resistentes a estos agentes. Hasta la fecha, los principales mecanismos implicados en esta resistencia han sido dos (ambos mediados por elementos cromosómicos): alteraciones en las dianas de las quinolonas y disminución en la acumulación del antibiótico en el interior bacteriano por impermeabilización de la membrana (pérdida de porinas o alteraciones del lipopolisacárido) o por expresión de sistemas de expulsión activa. En 1998, se describe por primera vez resistencia a quinolonas transmisible horizontalmente6. El gen qnr es el responsable genético de la resistencia y se encuentra dentro de un elemento móvil. La diseminación horizontal de mecanismos de resistencia a fluoroquinolonas abre la posibilidad de una rápida expansión de la resistencia a estos antimicrobianos, tanto en patógenos de animales como en seres humanos, más aún con el extenso uso que se hace de las mismas.
Mecanismo de acción y resistencia cromosómica
Las fluoroquinolonas inhiben la síntesis de ADN. La inhibición ocurre por la interacción del antibiótico con el complejo formado por la unión del ADN a las dianas de las quinolonas, la ADN-girasa y la topoisomerasa IV. Estas dos enzimas están estructuralmente relacionadas y constan de dos pares de subunidades diferentes, las subunidades GyrA y GyrB en el caso de la ADN-girasa, y las subunidades ParC y ParE en el caso de la topoisomerasa IV. Ambas enzimas son topoisomerasas tipo II, es decir, actúan cortando ambas hebras de un segmento de ADN, pasando otro segmento de ADN a través de la rotura, y volviendo a unir los extremos libres (fig. 1). La ADN-girasa está implicada en la relajación o enrollamiento del ADN, por ejemplo, durante el proceso de síntesis de ADN. La topoisomerasa IV está implicada en la separación de los cromosomas hijos tras la replicación del ADN7. En ambos casos, las fluoroquinolonas actúan atrapando a la enzima sobre el ADN durante la reacción de topoisomerización, cuando la enzima ha producido la rotura del ADN y ha generado extremos libres en éste. La unión del antimicrobiano estabiliza el complejo produciéndose una barrera física para el movimiento de la horquilla de replicación8, la ARN polimerasa9 y la ADN helicasa10. La colisión de este complejo con la horquilla de replicación desencadena una serie de sucesos, entre los que se incluyen la activación del sistema SOS, pero en general poco conocidos, que tienen como resultado final la muerte celular.
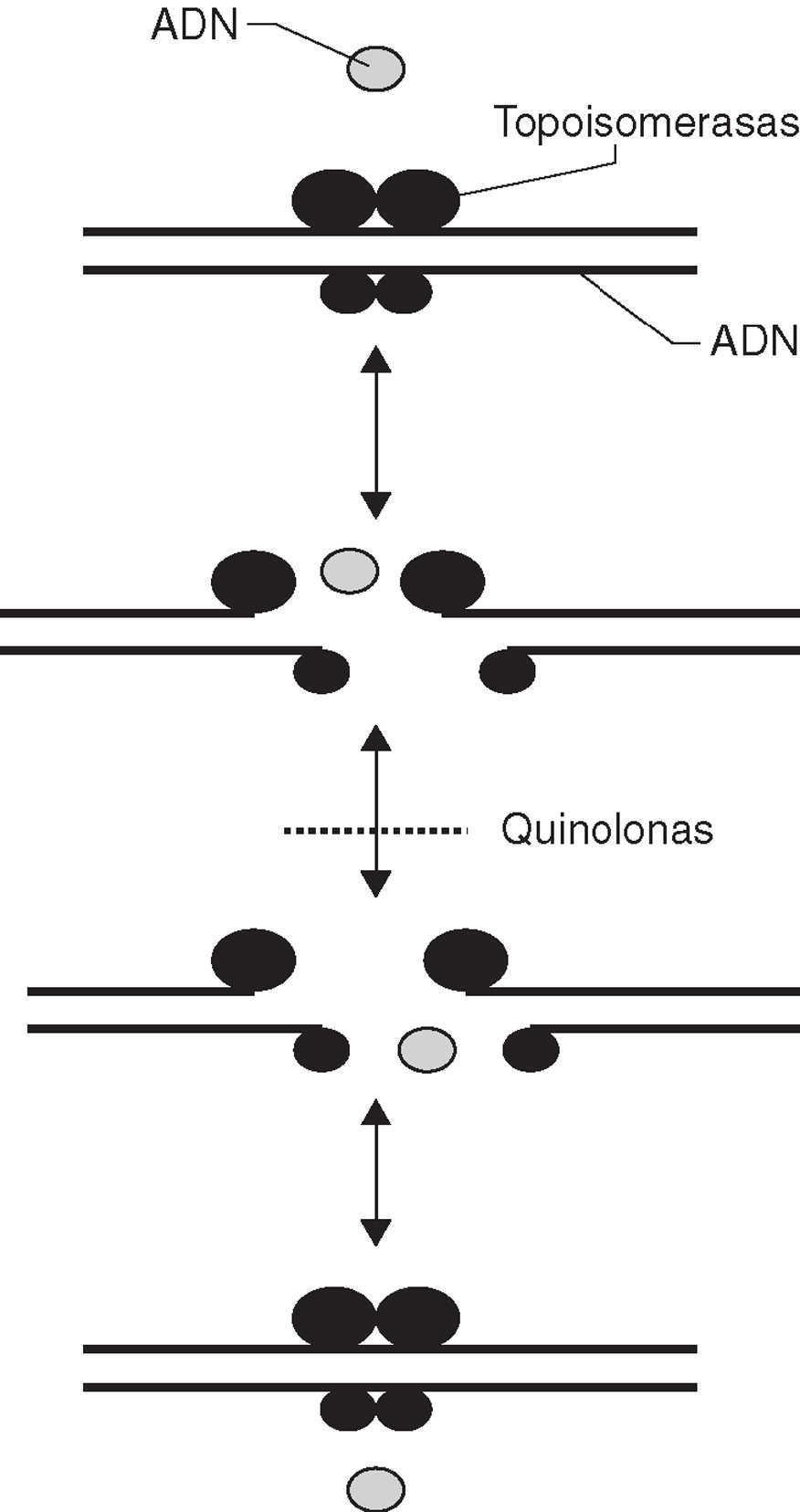
Figura 1. Mecanismo de acción de las topoisomerasas tipo II.
La diana de las quinolonas depende del compuesto considerado, y puede diferir entre grampositivos y gramnegativos. En los grampositivos la diana primaria es, en muchos casos, la topoisomerasa IV, mientras que en los gramnegativos es la ADN-girasa. Las fluoroquinolonas desarrolladas más recientemente, como moxifloxacino y clinafloxacino, presentan una afinidad similar por ambas dianas11.
En resumen, las quinolonas actúan en cuatro etapas: a) paso a través de las porinas de la pared bacteriana y b) de la membrana citoplasmática hasta llegar al citoplasma; c) inhibición de la ADN-girasa y/o topoisomerasa IV, y d) inducción de la respuesta SOS (fig. 2).
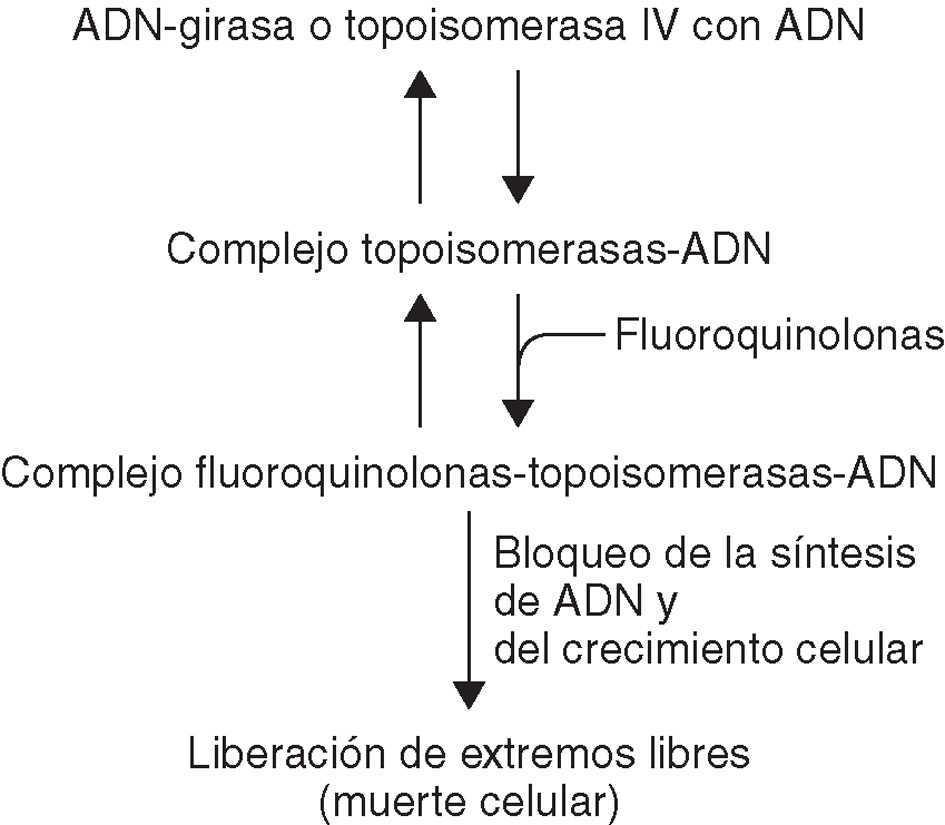
Figura 2. Interrupción de la acción de las topoisomerasas por quinolonas.
En su mayoría, la resistencia a quinolonas en bacterias gramnegativas se produce por mutaciones cromosómicas4,5,7. En Escherichia coli la modificación de la diana es determinada por mutaciones en gyrA o parC (genes que codifican para la subunidad A de la ADN-girasa y la topoisomerasa IV, respectivamente), y en menor medida en gyrB o parE (genes que codifican para la subunidad B de la ADN-girasa y la topoisomerasa IV, respectivamente). Estudios cristalográficos sobre la estructura de GyrA sugieren que los cambios aminoacídicos ocurren en la región que contiene el sitio activo, donde la enzima se une al ADN e interacciona con las quinolonas12 (tabla 1). Tanto en E. coli como en Staphylococcus aureus se ha demostrado que diferentes niveles de resistencia a quinolonas dependen de si las alteraciones ocurren en la diana primaria, la secundaria, o en ambas.

Otro factor importante para la resistencia a fluoroquinolonas se debe a mutaciones en bombas de transporte activo que expulsan compuestos tóxicos debido a su expresión o sobreexpresión provocando resistencia a diferentes grupos de antibióticos al mismo tiempo. Estos sistemas parecen estar en muchas, si no en todas las bacterias. En algunos microorganismos, como Stenotrophomonas maltophilia, este mecanismo puede tener una gran relevancia13. El gen norA codifica una bomba que contribuye a resistencia en S. aureus14, y acrAB codifica en E. coli un sistema de expulsión multidroga que asociado al producto de tolC modula la resistencia a quinolonas en esta especie15. La sobreexpresión de NorA, por una mutación en el promotor, provoca un aumento de 2 a 4 veces la concentración inhibitoria mínima (CIM) de ciprofloxacino16. Parece que la simple expresión de bombas de expulsión activa tiene un efecto limitado sobre la resistencia a fluoroquinolonas, pero su expresión basal sí contribuye de modo notable a la resistencia causada por otros mecanismos. Por ejemplo, si en cepas con mutaciones en gyrA se muta acrB, el nivel de resistencia disminuye considerablemente. Por otra parte, se han caracterizado un alto número de bombas potencialmente implicadas en la resistencia, con lo cual es difícil definir con exactitud el papel de cada una de ellas (tabla 2).

La pérdida de permeabilidad de membrana es otra de las formas de resistencia, por mutaciones en genes estructurales o reguladores que hace que disminuya el número efectivo de porinas (en E. coli genes ompC y ompF, en Klebsiella pneumoniae genes ompK35 y ompK36).
Recientemente se han comunicado, por primera vez, bajos niveles de resistencia causados por la expresión reducida de la topoisomerasa IV en S. aureus. Niveles reducidos de ParE son compatibles con la supervivencia bacteriana, aunque debe suponer un coste sobre la velocidad de división celular. Parece que este fenómeno debe llevar asociado otros mecanismos compensatorios que no revierten el fenotipo de resistencia17.
Mecanismos potenciales de resistencia a quinolonas mediada por plásmidos
En principio, el carácter hereditario de las mutaciones cromosómicas que causan resistencia a quinolonas haría que estos mutantes no requiriesen mecanismos de resistencia transferibles horizontalmente, ya que su supervivencia y diseminación vertical están aseguradas. Así, al ser la cepa receptora resistente a quinolonas no tendría ninguna necesidad de que se le transfiriese otro marcador de resistencia.
Plásmidos con genes cromosómicos
Una posibilidad con la que se especuló fue la adquisición de genes cromosómicos de la ADN-girasa o la topoisomerasa IV con mutaciones de resistencia a quinolonas por parte de plásmidos o elementos móviles. Además, la elevada tasa de resistencia en grampositivos (en particular en estafilococos), la demostración de la transferencia natural de material genético de grampositivo a gramnegativo18 y la ausencia de barrera para la expresión de genes de grampositivos en gramnegativos19 abre una vía teórica de resistencia transferible a quinolonas. En muchas especies la presencia de una topoisomerasa mutada es recesiva frente a la topoisomerasa silvestre original del microorganismo.
Inactivación de la droga
La inactivación de la droga (por oxidación, reducción, esterificación u otras reacciones) es el mecanismo de resistencia más común entre los patógenos bacterianos, produciendo resistencia cruzada a diferentes antimicrobianos del mismo grupo. Hasta el momento sólo se han descrito hongos capaces de degradar las quinolonas20,21. Puesto que estos compuestos son productos de síntesis en el laboratorio y no se producen por bacterias u hongos, es difícil pensar que existiese un proceso natural de presión hacia la resistencia a quinolonas en microorganismos ambientales o patógenos. Sí podría ocurrir, sin embargo, que las quinolonas se inactivaran por enzimas implicadas en la degradación de otros compuestos más o menos relacionados, y para los que sí existiese presión ambiental.
Mecanismo de acción de qnr
En 1987 se informó de resistencia a quinolonas mediada por plásmidos en una cepa de Shigella dysenteriae22, que más tarde no pudo ser comprobada. En 1998 se publicó por primera vez la existencia de una cepa clínica de K. pneumoniae aislada de un cultivo de orina recogido en Birmingham, Alabama (EE.UU.), que contenía un plásmido con alto rango de hospedador, y cuyos transconjugantes en E. coli incrementaban la resistencia a ácido nalidíxico de 4 a 32 mg/l, y a ciprofloxacino de 0,008 a 0,25 mg/l23. Este plásmido, denominado pMG252, aumenta de 4 a 8 veces la resistencia debida a mutaciones definidas en la ADN-girasa, porinas o bombas de expulsión activa24 y facilita la selección de mutantes resistentes a quinolonas, por mecanismos que aún no se conocen. La presencia de este plásmido no alteraba el patrón de expresión de porinas en el hospedador ni reducía la acumulación de quinolonas, lo que sugirió la existencia de un nuevo mecanismo de resistencia.
Para aclarar este posible nuevo mecanismo, el gen qnr de pMG252 se clonó, secuenció y después se amplificó por reacción en cadena de la polimerasa (PCR) y se introdujo en un vector de expresión. De este modo se pudo purificar la proteína codificada por qnr y estudiar su interacción con las quinolonas y sus dianas (ADN-girasa y topoisomerasa IV) mediante estudios de movilidad electroforética. Ello permitió demostrar que, al menos in vitro, Qnr protege a la ADN-girasa de E. coli de la inhibición por ciprofloxacino25. Esta protección es proporcional a la concentración de Qnr, e inversamente proporcional a la concentración de ciprofloxacino25. La topoisomerasa IV, diana secundaria de las quinolonas en E. coli, parece también ser protegida de las quinolonas por Qnr26.
Qnr pertenece a la familia de los pentapéptidos repetidos, de la que hasta el momento se conocen más de 90 miembros. Esta familia se define por la presencia de repeticiones en tándem de motivos A(D/N)LXX, donde X es cualquier aminoácido27. Estas proteínas se han encontrado en muchos géneros bacterianos, pero parece común en cianobacterias, pudiendo ser tanto proteínas de membrana como citoplasmáticas. Estas proteínas presentan estructura en a -hélice en su circunferencia externa y hojas b paralelas en su circunferencia interna28, una estructura apropiada para la interacción entre proteínas.
En esta familia de pentapéptidos existen dos miembros de especial relevancia en cuanto a la resistencia a quinolonas. El primero es McbG, una proteína que protege a las bacterias que sintetizan microcina B17 (MccB17) de su propia inhibición. MccB17 es un péptido modificado postrascripcionalmente de 3,1 kDa que bloquea la replicación del ADN29, y que puede, como el ciprofloxacino, inhibir la acción de la ADN-girasa30 y estabilizar el complejo ADN-ADN-girasa en presencia de trifosfato de adenosina (ATP)31 y de extremos libres de ADN. El mecanismo de autoinmunidad conferido por mcbG implica a otros genes: mcbE y mcbF, relacionados con el bombeo de MccB17 fuera de la célula32. Se ha comprobado que un plásmido que lleve el operón mcbEFG produce un aumento en la CIM de quinolonas de 2 a 8 veces33. En relación con este sistema, en el año 2002 se describió una nueva proteína (SbmC), que también protege a E. coli de la acción de MccB1734.
En E. coli, el operón mcb (responsable de la producción de MccB17) y el operón emr (que codifica la bomba EmrAB de resistencia a múltiples compuestos) comparten un mismo represor: EmrR. Además, los compuestos que inducen el operón emr reprimen el operón mcb33.
El segundo miembro de la familia de los pentapéptidos es MfpA, una proteína que se clonó del genoma de Mycobacterium smegmatis a partir de estudios sobre bombas de expulsión activa que contribuyen a la resistencia a quinolonas35. Los plásmidos artificiales que codifican MfpA incrementan la resistencia a ciprofloxacino hasta 4 veces. El mecanismo de resistencia no ha sido aún establecido, pero se sabe que MfpA no tiene efecto sobre la acumulación de ciprofloxacino marcado con C14.
La relación de los miembros de esta familia y Qnr es difícil de establecer, entre otras razones porque el porcentaje de homología entre Qnr y McbG o MfpA es del 19,6% y 18,9%, respectivamente25. Con los datos existentes sólo podemos especular con la posibilidad de que Qnr apareciese a partir de alguna proteína de inmunidad diseñada para proteger a la ADN-girasa de inhibidores naturales, o desde algún gen cromosómico de función desconocida que codificase una proteína de la familia de los pentapéptidos desde micobacterias, cianobacterias u otros grupos bacterianos.
Entorno genético deqnr
El gen qnr se encuentra, en las cepas en las que se ha descrito, en plásmidos transmisibles por conjugación. Estudios realizados con el plásmido en el que se describió por primera vez, pMG252, revelan que qnr se encuentra en un plásmido con amplio rango de hospedador transmisible por conjugación en especies como K. pneumoniae, E. coli, C. freundii, S. typhimurium o P. aeruginosa36. El qnr se localiza en el plásmido original de donde se aisló formando parte de una secuencia nucleotídica característica originalmente de los integrones In6 (del plásmido pSa) e In7 (de pDGO100)37, sugiriendo su presencia en un integrón de clase 125. Estos integrones poseen una región conservada común hacia 39 que contiene el gen qacEd1 (que confiere bajo nivel de resistencias a ciertos compuestos amónicos)38 y sulI (que confiere bajo nivel de resistencia a sulfamidas, pero que no se expresa en integrones al haber perdido su promotor)39. Un número inusual de integrones de clase 1 que contienen la región común de In6 e In7 llevan un elemento inicialmente denominado orf341, y ahora orf513, el cual se postula que codifica una recombinasa específica de sitio para la adquisición de genes de resistencia39. Muchos genes de resistencia, como los que codifican betalactamasas plasmídicas, se encuentran localizados dentro de elementos móviles de este tipo, y también dentro de transponsones, lo cual, como se sabe, potencia su diseminación40. En estos casetes de resistencia, incluido qnr, el elemento de 59 pb41 se ha perdido, indicando que, efectivamente, orf513 debe estar implicado en la adquisición específica de sitio de genes.
Es importante señalar que existe una relación estadísticamente significativa entre la resistencia a quinolonas y betalactámicos42, y sea circunstancial o no, pMG252 contiene la betalactamasa de espectro ampliado FOX-525. Así, se abre una vía de corresistencia a dos familias muy importantes de antimicrobianos, sobre todo en cepas con algún mecanismo de resistencia a quinolonas como la pérdida de porinas, donde la presencia de qnr facilita alcanzar altos niveles de resistencia.
En un estudio más reciente43, se analizó el entorno genético de los transconjugantes de dos cepas de E. coli de un hospital de Shangai resistentes a quinolonas en las que se había identificado la presencia de qnr. En estas cepas se halló que qnr se encuentra formando parte de un integrón de clase 1, perteneciente a la familia In4, adyacente a orf513 y aguas arriba de ampR, qacEd1 y sul1 (fig. 3). El gen qnr del plásmido original pMG252 tiene una localización similar, pero en los plásmidos de las cepas de Shangai, el gen ampR se encuentra inmediatamente aguas debajo de qnr, mientras que en pMG252, qacEd1 y suestán directamente aguas debajo deqnr. En estos dos nuevos plásmidos, los integrones que contienen qnr, In36 e In3743, presentaban una estructura similar a la de pSal-144, pero ampC se sustituye por qnr (fig. 3). En el resto de cepas qnr positivas del estudio de las cepas de Shangai se comprobó por PCR que ampR se encuentra próximo a orf513, pero ampC ha sido sustituido por qnr.
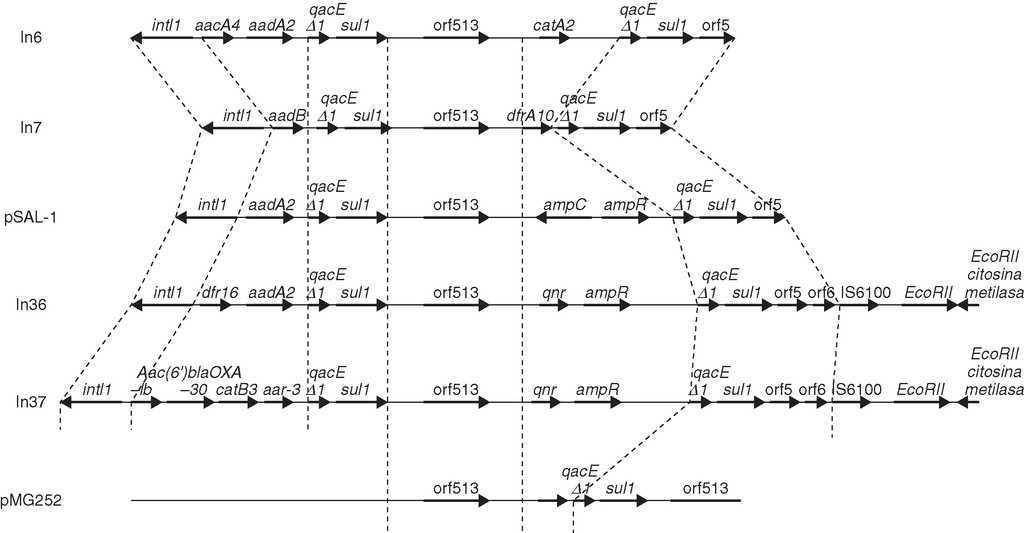
Figura 3. Entorno genético de cepas en las que se ha encontrado qnr y comparación de la secuencia con otros integrones de clase 1.
En muchas ocasiones, la movilidad de los integrones se debe a su presencia dentro de transponsones o plásmidos. Los integrones que llevan qnr parece que son móviles entre diferentes plásmidos. Esta interpretación se apoya en varias observaciones: en primer lugar, qnr se halla en plásmidos de diferentes tamaños en las cepas clínicas donde se encontró; en segundo lugar, en una de las cepas se encuentra en dos plásmidos de diferente tamaño (además, estos plásmidos son capaces de transferir qnr por conjugación)43; en tercer lugar, en algunos transconjugantes la señal de hibridación de qnr se pierde, sin que se pierda la presencia del plásmido cuando se crecen a elevada temperatura o en ausencia de presión antibiótica43. El mecanismo por el que los integrones se movilizan no está claro, pero esta movilidad se apoya en sus múltiples localizaciones, que indican movimientos en el pasado, y por la presencia en algunos de ellos de repeticiones directas de 5 pb que son consistentes con movimientos por algún mecanismo de transposición45. Parece razonable pensar que los integrones de clase 1 pueden ser movilizados si poseen las secuencias características IRi e IRt, y los genes tni que codifican enzimas de transposición son aportados en trans desde otras estructuras, ya sean plasmídicas o cromosómicas45. Los dos integrones descritos que contienen qnr, In36 e In37, contienen tanto las secuencias IRi e IRt como las duplicaciones directas de 5 pb, indicando que es posible el movimiento por transposición43. La movilidad de qnr podría ser posibilitada por la presencia de genes tni en otro transponsón en estas cepas clínicas.
Efecto de Qnr sobre la actividad de las quinolonas
En la tabla 3 se pueden observar las CIM de diferentes quinolonas frente a algunas de las cepas clínicas publicadas y de transconjugantes obtenidas a partir de ellas. Como puede observarse, las CIM de ciprofloxacino frente a los transconjugantes están entre 0,125 y 2 mg/l, lo cual representa un aumento de 16 a 250 veces la CIM, en relación con la del recipiente. En un trabajo reciente46 se probó la actividad de diferentes quinolonas frente a Qnr, y se vio que antimicrobianos como sitafloxacino, BAYy3118 y premafloxacino son más activos que ciprofloxacino, tanto en los transconjugantes como en los donadores.
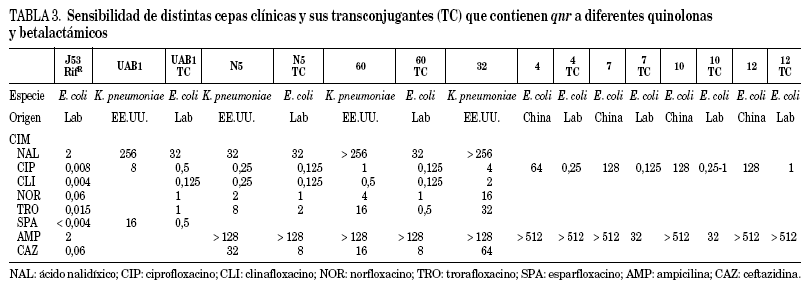
Hay que señalar que la presencia de qnr en los transconjugantes estudiados es estable tras sucesivos pases en medios sin presión antibiótica, permaneciendo el fenotipo de resistencia a quinolonas, acompañado de otras resistencias, excepto en un caso donde se perdió la resistencia a quinolonas, y también la asociada a trimetoprima+sulfametoxazol, cloranfenicol y tetraciclina43. El plásmido que poseía este transconjugante en particular tenía un tamaño ligeramente inferior al esperado, indicando que al menos en este caso parte del integrón se pudo perder durante el proceso de conjugación43.
Se ha observado que la resistencia a quinolonas mediada por plásmido, aunque produce resistencia de bajo nivel, también facilita la selección de resistencia de alto nivel a quinolonas47. Se sabe desde hace tiempo que los plásmidos que median resistencia a bajas concentraciones de estreptomicina incrementan la selección de niveles mayores de resistencia48. Así, cuando qnr se expresa en una cepa deficiente en porinas, las CIM para ciprofloxacino, levofloxacino y moxifloxacino aumentan de 8 a 32 veces49, pasando las CIM de estas quinolonas de 0,25-0,5 a 4-8 mg/l. Existe un efecto aditivo de los distintos mecanismos de resistencia a quinolonas con la presencia de qnr50. Se piensa que la baja resistencia a un antimicrobiano permite a las poblaciones bacterianas alcanzar concentraciones a las cuales la aparición de mutaciones secundarias que permiten la aparición de alta resistencia puede ocurrir. Esto sería lo que ocurriría con la resistencia a estreptomicina y a quinolonas codificada por el plásmido original donde se describió qnr, pMG252, donde la frecuencia de mutaciones de alta resistencia aumenta en los transconjugante respecto del recipiente E. coli J53 sin plásmido47.
Prevalencia de Qnr
Los integrones son elementos comunes en los aislados clínicos bacterianos en bacterias gramnegativas, estando presentes hasta en más del 40% de éstas. Además, los aislados que contienen estos elementos son significativamente más resistentes a antibióticos como quinolonas, aminoglucósidos y betalactámicos51, sugiriendo la posibilidad de que qnr u otro gen relacionado esté presente en cepas de este tipo, muchas de ellas con secuencias conservadas de integrones de clase 1.
Aún no se han realizado suficientes estudios para determinar la prevalencia real del determinante de resistencia a quinolonas qnr transferible horizontalmente. En un primer trabajo52, de un total de 350 cepas de 13 géneros diferentes de bacterias gramnegativas en su mayoría procedentes de Estados Unidos, sólo presentaron el gen qnr un aislado de E. coli, cuatro aislados de K. pneumoniae y un aislado de Klebsiella sp. de cultivo de orina y de esputo. En un segundo trabajo43, sobre 78 cepas clínicas de E. coli resistentes a quinolonas recogidas de cinco hospitales de Shangai, China, entre marzo de 2000 y marzo de 2001, el 7,7%, es decir, 6 cepas, poseían el gen qnr. En este caso las cepas procedían del mismo hospital, pero de diferentes pacientes. Un tercer estudio53, sobre un total de 266 cepas de E. coli y 159 cepas de K. pneumoniae con diferentes fenotipos de resistencia a betalactámicos y quinolonas dio como resultado el hallazgo de tres cepas de K. pneumoniae que contenían el gen qnr. Por el contrario, ninguna de las cepas de E. coli fueron positivas. Recientemente, Wang et al54 han publicado un trabajo sobre cepas clínicas de K. pneumoniae y E. coli de diferentes estados de Estados Unidos en el que la frecuencia de qnr es mayor del 11% en K. pneumoniae (no se detectó en ninguna cepa de E. coli). Alguna de estas cepas producían SHV-7, siendo ésta la primera vez que se describe una asociación entre una betalactamasa de espectro extendido y Qnr en la misma cepa. De estos estudios se deduce que este gen está ampliamente distribuido en cepas clínicas resistentes a quinolonas de K. pneumoniae en Estados Unidos y E. coli del sudeste asiático.
Conclusiones y perspectivas
La identificación de qnr en cepas clínicas de K. pneumoniae productoras de betalactamasas plasmídicas aisladas en Estados Unidos, y su hallazgo en cepas de E. coli procedentes del sudeste asiático indican la emergencia de este nuevo mecanismo de resistencia a quinolonas en cepas clínicas. Además, la posibilidad de su diseminación a otros géneros de bacterias gramnegativas podría agravar el problema; qnr media la resistencia a quinolonas desde integrones de clase 1, unido a otros genes de resistencia, lo cual tiene un efecto adicional, ya que la disminución de sensibilidad a quinolonas en presencia de otras resistencias promueve la selección de genes de este tipo que se encuentran en el mismo integrón. Son necesarios nuevos estudios que aclaren la prevalencia real de este mecanismo (incluyendo grampositivos), los mecanismos de resistencia y selección de mutantes en presencia de qnr, así como modelos animales para el estudio de la trascendencia clínica e in vivo.

