









The concept of aggressive pituitary tumor (APT) has been precisely defined in recent years. These tumors are characterized by morphological (radiological or histopathological) data of invasion, proliferative activity superior to that of typical adenomas and a clinical behavior characterized by resistance to standard therapies and frequent recurrences. The absence of cerebrospinal or distant metastases differentiates them from the pituitary carcinoma. APTs account for about 10% of all pituitary neoplasm. Proper diagnostic implies participation not only of radiological and hormonal investigation but also a thorough pathological assessment including proliferation markers and immunohistochemistry for hormones and transcription factors. Surgical resection, aiming gross total resection or tumor debulking, is the mainstay initial therapy in most patients. Most patients with APTs need more than one surgical intervention, pituitary radiation, sometimes on more than one occasion, and multiple sequential or combined medical treatments, to finally be doomed to unusual treatments, such as alkylating agents (temozolomide alone or in combination), molecular targeted therapies, or peptide receptor radionuclide therapy. Multimodal therapy, implemented by experts, preferably in specialized centers with high volume caseload, is the only way to improve the prognosis of patients with these uncommon tumors. The research needs in this area are multiple and include a greater knowledge of the molecular biology of these tumors, establishment of protocols for monitoring and sequencing of treatments, development of multicenter studies and international registries.
El concepto de tumor hipofisario agresivo (THA) se ha definido con más precisión en los últimos años. Son tumores caracterizados por signos morfológicos (radiológicos o histopatológicos) de invasión, actividad proliferativa superior a la de los adenomas típicos y un comportamiento clínico caracterizado por resistencia a los tratamientos habituales y recidivas frecuentes. La ausencia de metástasis cefalorraquídeas o a distancia los diferencia del carcinoma hipofisario. Los THA suponen alrededor del 10% de todas las neoplasias hipofisarias. Un diagnóstico apropiado exige no solo investigación radiológica y hormonal, sino también una valoración histopatológica detenida que incluya marcadores de proliferación e inmunohistoquímica para hormonas y factores de transcripción. La resección quirúrgica encaminada a la resección total o la reducción del volumen tumoral es el tratamiento inicial clave en la mayoría de los pacientes. La mayoría de los pacientes con THA necesitan más de una intervención quirúrgica, irradiación hipofisaria, a veces en más de una ocasión, y diversos tratamientos médicos consecutivos o combinados, y están predestinados a terminar recibiendo tratamiento inhabituales como fármacos alquilantes (temozolomida sola o en combinación), tratamientos multidiana o tratamientos con péptidos radiomarcados. El tratamiento multimodal aplicado por expertos, preferiblemente en centros especializados con gran volume de pacientes, es el único modo de mejorar el pronóstico de los pacientes con estos tumores poco frecuentes. Las necesidades de investigación en este campo son enormes, e incluyen la de un mayor conocimiento de la biología molecular de estos tumores, el establecimiento de protocolos de vigilancia y secuenciación de los tratamientos, el desarrollo de estudios multicéntricos y registros internacionales.
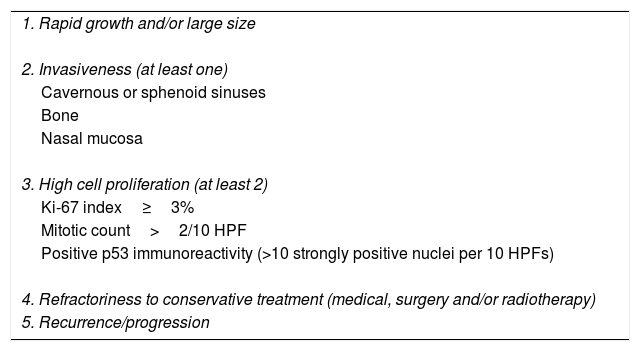
Pituitary tumors (PT) are the second brain tumor accounting for 15% of all intracranial neoplasms.1 Although in most cases they are benign tumors with an adequate response to conservative therapy, a small percentage are associated with criteria of aggressiveness and refractoriness to conservative treatment with medical therapy, surgery with or without radiotherapy being associated with high morbidity and mortality (up to 28%; median duration from initial diagnosis to death of 11 years) (Table 1).2–6
Main clinical and pathological criteria for aggressive pituitary tumors.
| 1. Rapid growth and/or large size |
| 2. Invasiveness (at least one) |
| Cavernous or sphenoid sinuses |
| Bone |
| Nasal mucosa |
| 3. High cell proliferation (at least 2) |
| Ki-67 index≥3% |
| Mitotic count>2/10 HPF |
| Positive p53 immunoreactivity (>10 strongly positive nuclei per 10 HPFs) |
| 4. Refractoriness to conservative treatment (medical, surgery and/or radiotherapy) |
| 5. Recurrence/progression |
Abbreviations: HPF, high power field.
Following the recent recommendations of the European Society of Endocrinology aggressive pituitary tumors (APT) should be managed by a multidisciplinary team.6 We herein review the most recent and novel data related to the different therapeutic options and their clinical outcomes in APT from the point of view of several medical and surgical specialties. In this review, pituitary carcinoma, defined as pituitary tumors with cerebrospinal and/or systemic metastasis, will not be considered.
Definition of aggressive pituitary tumorAPT are a rare entity that should be considered as a tumor with malignant potential.7 These neoplasias are characterized by rapid growth and usually large tumor size, invasion of adjacent structures, an aggressive clinical behavior with poor response to conventional treatment (medical therapy, surgery±radiotherapy), high rate of recurrence, and elevated morbidity and mortality.4,8–14
APT is not the same as an invasive pituitary adenoma (PA). Although most APTs are invasive, that is, show radiological or pathological signs of invasion to the cavernous or sphenoid sinuses, bone, or nasal mucosa, some invasive PAs do not behave like APT.11 On the other hand, although APT usually present with histological markers of increased proliferation such as Ki-67 index >3%, elevated mitotic count, and/or positive p53 expression, the presence of these proliferation markers are not essential to predict the aggressive behavior of all PAs.6 However, it has been reported that the coexistence of an invasive PA with histological markers of cell proliferation increases the probability of developing an aggressive tumor.15,16 Therefore, we suggest that to establish the diagnosis of APT it would be appropriate to consider at least 3 of the 5 criteria shown in Table 1.
Prevalence and demographic characteristicsThe lack of a clear definition and standardized criteria on the definition of APT in the last years has contributed to the absence of epidemiological studies related to this subtype of tumors. It has been estimated a prevalence of APT ranging between 2.5% and 10% of all pituitary adenomas according to surgical series.5,6,9,16
A recent European Society of Endocrinology (ESE) survey of a cohort of 125 patients with APT showed a mean age at diagnosis of 43 years (range 4–79 years), with a predominance in males (64.5% vs. 35.5%), and higher prevalence of functioning versus non-functioning tumors (64.8% vs. 35.2%). Mean pituitary surgeries was 2.7 pituitary surgeries and 1.2 courses of radiotherapy. The more frequent histological subtype was corticotroph adenoma (44.8%), followed by prolactinoma (20%), null cell adenoma (16.8%), somatotroph adenoma (11.2%), gonadotroph adenoma (4%), and thyrotroph adenoma (3.2%).5 In this study APT showed a pattern of invasive growth in 87% of the patients, 70% of the tumors grew after radiotherapy or did so after 2 previous surgeries, and 54% had resistance to medical treatment.5
Histopathological characterizationIn the 2017 WHO classification, the term “atypical adenoma” is abandoned, and the term of high risk recurrence adenomas is incorporated. They are defined as those adenomas that show features that tend to predict recurrence and resistance to conventional therapy. These features include rapid growth, radiological invasion, and a high Ki-67 proliferation index.
Aggressive histological typesAccording to the World Health Organization (WHO) the main histological subtypes of PAs that usually show an aggressive behavior are silent corticotroph adenomas, lactotroph adenomas in males, sparsely granulated somatotroph adenomas (SGSA), Crooke cell adenomas, and plurihormonal positive PIT-1 (pituitary transcription factor 1) positive adenomas (previously called silent subtype 3 adenoma).17
Silent corticotroph adenomas are composed of faintly basophilic or chromophobic PAS positive cells with weak or patchy positivity for ACTH and for specific corticotroph lineage transcription factor (TPIT)-lineage adenohypophyseal cells.
Densely granulated lactotroph ademoma (DGLA) has an eosinophilic to acidophilic cytoplasm, with strong and diffuse PRL expression throughout the cytoplasm (Fig. 1) and co-express PIT1 and ER-alpha (estrogen receptor). Lactotroph macroadenoma in men is a rare and aggressive subtype of lactotroph adenoma.
![Histopathological study of an aggressive prolactinoma showing densely granulated lactotroph adenoma (A) with immunostaining positive for prolactin (B), with histological signs of aggressiveness [cellular pleomorphism and nuclear atypia, 2 mitotic figures for 10 high power fields (C, arrow), bone infiltration and Ki67 10% (D)].](https://static.elsevier.es/multimedia/25300164/0000006700000007/v1_202007210834/S2530016419302265/v1_202007210834/en/main.assets/gr1.jpeg?xkr=ue/ImdikoIMrsJoerZ+w997EogCnBdOOD93cPFbanNf23BlP/DWEpPOpuWlfofnyqxn4xVHcGY2cyFujnbUAmO/P5kgGivG0klIV0/1ajQruXtEj8JJdPZQHSRpKNHirHSw3924DMxwblqsUscxnH3jEycp5RS2oapHD3ubtz61d5tshIqGInPS+nAwJ32sKr7YihC9255nOhHgMGLgIEQostHcOQQV953nrp7+2aoEyEHTHLpiQD2VTI21kSjNocfD/bY5WPFv+ieiWPJzzueGlOfILpfTngb3tYaJuK/FY7Q2xS+OB5RhpRA2+x+Lu "Histopathological study of an aggressive prolactinoma showing densely granulated lactotroph adenoma (A) with immunostaining positive for prolactin (B), with histological signs of aggressiveness [cellular pleomorphism and nuclear atypia, 2 mitotic figures for 10 high power fields (C, arrow), bone infiltration and Ki67 10% (D)].")
Histopathological study of an aggressive prolactinoma showing densely granulated lactotroph adenoma (A) with immunostaining positive for prolactin (B), with histological signs of aggressiveness [cellular pleomorphism and nuclear atypia, 2 mitotic figures for 10 high power fields (C, arrow), bone infiltration and Ki67 10% (D)].
SGSA are composed of chomophobic pale eosinophilic tumor cells that are positive for PIT1. Nuclear pleomorphism, including multinucleated bizarre cells, can be noted. Consistent with sparse granularity, positivity for GH is variable, with reactivity ranging from weak to focal or patchy. These neoplasms are associated with poor response to SSA, larger tumor size, lower levels of GH and IGF1 and T2-hyperintensity on MRI.
Crooke cell adenomas are composed of tumor cells with Crooke hyaline change. Ring-like cytokeratin expression is typical of this neoplasm. ACTH expression is dislocated to the cell periphery and yuxtanuclear region. They show a clinically more aggressive behavior.
Lastly, plurihormonal PIT1 positive adenomas are chromophobic and they are variably positive for GH, PRL, TSH, alpha subunit, and ACTH, and show extensive nuclear PIT1 expression. They are aggressive in terms of their size, grow rate, and invasiveness, with cavernous sinus invasion, rate of persistent tumor and recurrence.18–21
Biomarkers of pituitary tumor aggressivenessThe most common alteration reported in APT is the allelic loss of the short arm of chromosome 11 (11p), mainly in lactotroph adenomas.22 The role of MYO5A, a member of the myosin family, in tumor cell invasion and metastasis has also been reported.23 Downregulation of miR-15a and miR-16-1 has been associated to tumor size in both corticotropinomas24 and somatotroph and lactotroph adenomas.25 In addition growth factors such as epidermal growth factor (EGF), vascular endothelial growth factor (VEGF), fibroblast growth factor (FGF) and their receptors (EGFR, VEGFR and FGFR, respectively) have also been involved in the aggressiveness of the pituitary tumors.26,27 Expression of several metalloproteinases (MMPs), such as MMP9 and MMP2 has been correlated with the degree of invasion and adenoma phenotype in some studies.28–30 Galectin-3 has been studied as a predictive marker of aggressive tumor behavior in corticotroph and lactotroph adenomas.31 Pituitary tumor-transforming gene (PTTG) was higher in hormone-secreting invasive PAs compared to noninvasive ones32 whereas a PTTG expression correlated with the proliferative activity and recurrence status in PAs.33 Other more recent studies have shown that insulin-like growth factor 1 receptor (IGF1R) expression is a more helpful molecular marker than PTTG in PA management, whereas, Ki-67 showed no association to tumor behavior.34 The reliability of genomic and molecular markers has yet to be evaluated in large prospective studies, alone or as part of multimodal prognostic models.
Medical therapyAggressive corticotroph adenomaCorticotroph adenomas or corticotropinomas are the most frequently pituitary tumors associated with aggressive behavior.5 Among them are silent corticotropic adenomas, mainly type 2 sparsely granulated tumors (Fig. 2), and Crooke cell adenomas.35
 at different times of the disease. A. Before second surgery. B. After second surgery. C. Four months after second surgery.")
Corticotropinomas express somatostatin receptor (SST), both subtype 2 (SST2) and subtype 5 (SST5).36 However, due to the hypercortisolism associated with Cushing's disease (CD) reduces the expression of SST2, the main SST in functioning corticotropinoma is SST5.37,38 It is because of that octreotide and lanreotide, SS analogs (SSA) with high affinity for SST2, have a limited effect on corticotroph adenomas. However, pasireotide, a multireceptor ligand SST analog with a high binding affinity for SST5 has shown its efficacy in CD with urinary free cortisol (UFC) normalization in 26% of patients.39 Moreover, long-acting once-monthly pasireotide has proven effective in CD patients normalizing UFC in about 40% of patients with persistent or recurrent disease after initial surgery.40 Little information on the effect of pasireotide on tumor size in CD is nowadays available, although some studies have reported significant tumor shrinkage in 62.5% of patients after 6 months and in 100% of patients after 12 months, with occasionally radiological disappearance of the tumor.41 To our knowledge, the effect of pasireotide on tumor volume in aggressive corticotropinomas is not really known.
Pasireotide has been accompanied by a reduction in plasma ACTH concentrations in patients with Nelson syndrome, an invasive corticotroph tumor that develops after bilateral adrenalectomy in CD42,43; however, the effect on tumor volume is less clear. While some authors describe a reduction in tumor size,42 others did not found any significant effect after 28-week of therapy.43
Corticotropinomas express functional dopamine receptor type 2 (D2R) in approximately 80% of patients.44,45 In fact, it has been reported a normalization in cortisol secretion up to 20–40% of CD treated with cabergoline.44,46 However, the role of dopamine agonists (DA) on corticotropin secretion and tumor volume in aggressive corticotropinomas has not been fully elucidated. The expression of both SST5 and D2R in corticotropinomas would support the combined therapy with cabergoline and pasireotide in aggressive corticotropinomas.
Temozolomide (TMZ), an oral imidazotetrazine second-generation DNA alkylating agent which causes methylation at the O6-position of guanine and alkylation at the N7-positions has shown antitumor activity against high-grade tumors including high-grade glioma. Since 2006, TMZ has become a therapeutic alternative in APT refractory to conventional therapy with medical treatment, surgery with or without radiotherapy. Nowadays, TMZ is considered the first-line therapy for ATP following documented tumor growth.6
TMZ has been associated with a positive response in aggressive corticotropinomas.47,48 The overall response rates to TMZ in corticotropinomasis around 60%.49 Similarly, TMZ has been shown as an effective therapeutic alternative in invasive adenomas in Nelson's syndrome.50 TMZ has also been shown to be effective for aggressive corticotropinoma in both children and in the elderly.6,51
The efficacy of TMZ therapy has been related to the tumor expression of O6-methylguanine-DNA-methyltranferase (MGMT), a DNA repair protein. A low MGMT expression assessed by immunohistochemistry has been related to a better therapeutic response to TMZ,5,6,52 although not in all cases.51,53,54 Other authors have reported that tumoral MGMT content also predicts survival in APT patients.55 Another predictive marker of TMZ response is the expression of DNA mismatch repair protein (MSH6).56,57 In clinically aggressive corticotropinomas a low or absent MGMT expression has been associated with a clinical therapeutic response.47
The standard dose of TMZ used in aggressive corticotropinoma is usually 150–200mg/m2/day during 5 days every 28 days. The number of cycles is variable, varying between 4 and 24 cycles. Patients should be re-evaluated by imaging (MRI in most instances) after the 3rd cycle and, in the case of tumor progression, treatment should be suspended. Other reasons for withdrawing the drug would be severe side effects, such as intense fatigue, nausea/vomiting, and cytopenias (thrombocytopenia and/or leukopenia). A life-long follow-up with hormonal and imaging studies every 3/12 months according to the clinical evolution is recommended.6
Although treatment with TMZ in aggressive corticotropinoma is effective and safe, this therapy is not always successful after tumor progression following response to TMZ.58 However, a second trial of 3 cycles of TMZ has been suggested.6 These patients can also benefit from combined therapy with TMZ and capecitabine (CAPTEM therapy). This regimen can achieve a high therapeutic response rate and prolonged survival, even with radiographic complete remission in some cases.59 Moreover, a low MGMT expression and adequate levels of mismatch repair enzymes (MLH-1, MSH-2, MSH-6, and PMS-2) seem to be important for the efficacy of this therapy.56,57 Other therapeutic option for patients with rapid tumor growth is the combination of TMZ with radiotherapy.6 For those patients with rapid tumor progression on TMZ treatment a trial with other systemic cytotoxic therapy has also been recommended. Among other possible therapeutic alternatives are targeted therapies, such as Raf/MEK/ERK and PI3K/Akt/mTOR pathways, tyrosine kinase inhibitors targeting the VEGFR, and VEGF-targeted therapy (bevacizumab).6 A suggested therapeutic approach is shown in Fig. 3.
Aggressive lactotroph adenoma
Lactotroph adenomas or prolactinomas are usually benign tumors sensitive to conventional therapies, including medical therapy, surgery, and radiotherapy. However, some of them demonstrate aggressive behavior, mainly those densely granulated and acidophil stem cell adenomas, characterized by large size, accelerated growth, high recurrence rate, and persistent growth despite successive therapies.60
DA therapy, mainly cabergoline, is the first-line therapy in prolactinomas due to it has clearly demonstrated its efficacy in the control of hyperprolactinemia and tumor volume.61 These benefits have been observed not only in micro-, but also in macroprolactinomas. DA therapy is effective even in giant prolactinomas (≥4cm) in whom normoprolactinemia is achieved in 60% and reduction (≥30%) in tumor size in 83%. However, hormonal resistance to DA (absence of normoprolactinemia after bromoctriptine ≥15mg/day or cabergoline ≥2.0mg/week) has been reported in up to 10% of prolactinomas and to 25% of the most aggressive prolactinomas.13,61–63 Before surgery, the therapeutic alternatives in prolactinomas resistant to DA are change of the drug and increase the dose until reaching a greater therapeutic response with adequate tolerance.64 The increase in dose of cabergoline (up to 11mg/week) has been shown to be effective in controlling hyperprolactinemia in most resistant patients.65 Therefore, it has been proposed to use the maximum tolerable dose in patients with aggressive prolactinoma.6
It is known that prolactinomas express different SST subtypes, mainly SST5 and SST1, being SST2 expression low.66,67 Studies in prolactinoma cell cultures have shown that SST5 agonists are more effective than SST2 agonists in suppressing PRL secretion, although less effective than DA. In fact, pasireotide can suppress PRL secretion in most prolactinomas in vitro probably due to its high SST5 affinity.68 Some case reports have shown excellent response to pasireotide long-acting release (PAS-LAR) therapy in an aggressive and dopamine-resistant prolactinomas suggesting this therapy as a new potential treatment option before starting TMZ.69–71 Moreover, PAS-LAR therapy seems to induce cystic degeneration, tumor cell necrosis, or both in prolactinomas.69
On the other hand, a SST2 overexpression in prolactinomas resistant to DA therapy has also been reported.72 However, the induced octreotide SST2-mediated PRL suppression seems to be also lower than that induced by DA. In 2011, an isolated case report with aggressive DA-resistant macroprolactinoma showed a positive uptake in the scintigraphy with 111In-pentetreotide indicating the presence of functioning SST on tumor tissue. This patient was treated after surgery with combined treatment adjuvant with cabergoline plus octreotide, achieving an adequate degree of control of hyperprolactinemia and tumor size after 2 years of treatment.73 It has been suggested a potential additive effect induced by the combined therapy with cabergoline and octreotide,73 as reported in human prolactinomas in vitro studies.74 Long-term therapeutic success with multimodal medical therapy (cabergoline, lanreotide, and TMZ) has also be reported in aggressive prolactinoma.75
It has been reported an activation in mTOR pathway in prolactinomas. In fact, everolimus have shown antiproliferative actions in vitro, suggesting this drug as a novel therapeutic option for some aggressive PRL-secreting tumors unresponsive to conventional therapy.76
Conventional chemotherapy with drugs such as fluorouracil, nitrosoureas, and carboplatin has shown little therapeutic effect in the management of aggressive prolactinoma. TMZ has proven its efficacy as salvage therapy in some, but not all patients,53 with refractory, recurrent, and invasive prolactinomas achieving a significant tumor shrinkage and reduced PRL secretion.75,77–82 The overall response rates to TMZ in prolactinomas is around 67%.49 TMZ was accompanied by an 80% reduction in tumor volume with a normalization of serum prolactin concentrations in a 60-year-old male with aggressive prolactinoma after 12 cycles of TMZ.77 One year later, Losa et al., 201078 reported two other patients treated with 12 cycles of TMZ. In one of them, serum PRL decreased significantly with stable tumor response, while in the other one, normoprolactinemia and partial response of tumor size were achieved. In other series of APT patients, no hormonal response or reduction in tumor size was found in one aggressive prolactinoma patient.53 A series of 13 aggressive prolactinomas, hormonal and/or tumor response was reported in 7 of them (53.8%).79 More recently a patient survey developed by the task force on APT appointed by the European Society of Endocrinology reported a complete response (CR) in 5%, partial response (PR) in 45%, stable disease (SD) in 26%, and progression (P) in 24% of patients in a group of 38 aggressive lactotroph tumors.5 These results have made this drug be recommended as the first-line chemotherapy for aggressive prolactinomas after failure of standard therapies.6 TMZ has also been shown to be effective not only in adults with aggressive prolactinomas, but also in children83 and in the elderly.51
The immunopositivity of MSH6 has been positively correlated with TMZ response, suggesting that the preservation of MSH6 function can contribute to the effectiveness of TMZ in aggressive prolactinomas.84
TMZ is able to achieve prolonged periods of tumor remission of up to 6 years after its withdrawal allowing to reduce the dose of DA.82 On the other hand, retreatment with TMZ has also shown a rapid (after 4th cycle) biochemical and radiographic response in a recurrent aggressive prolactin-secreting PA.85 Lastly, TMZ therapy seems to be effective when combined with radiotherapy or another chemotherapeutic agent. In fact, TMZ plus radiotherapy was associated with a significant better response rate compared to TMZ alone in aggressive prolactinomas.5 TMZ in combination with capecitabine, bevacizumab, and thalidomide has been accompanied by PR and SD in isolated cases.5Fig. 3 shows a therapeutic approach for aggressive lactotroph adenoma.
Aggressive somatotroph adenomaFirst generation SSA, octreotide and lanreotide, have high affinity for SST2 and less for SST5, and activate the signaling pathway that inhibits GH production. About 40–50% of acromegalic patients exhibit incomplete response to SSA and 10% are resistant to these drugs.37,86 Although these compounds are clearly less effective in invasive macroadenomas than in microadenomas, standard therapy with octreotide or lanreotide are still considered the first-line medical therapy for aggressive somatotropinomas due to their effects both on GH secretion and tumor mass.87,88
In a retrospective analysis of 34 patients with giant adenomas, many of them with aggressive behavior, the treatment with first generation SSA achieved remission of the disease in 6 patients and partial control (IGF1 <1.5× upper limit of normality [ULN]) in 9. This remission rate was considered far below the rate considered appropriate for patients harboring GH secreting macroadenoma.89
Different histological, molecular and genetic factors that contribute to resistance to SSA in some pituitary tumors have been reported, such as poorly granulated tumors, larger tumor size, decrease in SST density, receptor mutations, diverse expression of subtypes of SST, expression of truncated isoforms of SST5, mutation in the aryl hydrocarbon receptor-interacting protein (AIP) gene, or deletion of exon 3 of GH receptor.90–96 Therefore, research into new drugs that improve the effectiveness of these first generation agents has been carried out in recent years.
A recently reported large multicenter, randomized, 12-month, head-to-head superiority study investigated the efficacy and safety of pasireotide LAR compared with octreotide LAR in patients with denovo acromegaly and in those who had under-gone unsuccessful surgery.97 In this study, 31.3% of patients treated with pasireotide LAR, but only 19.2% of those treated with octreotide LAR, achieved levels of GH <2.5μg/l and age-normalized levels of IGF-1. Pasireotide LAR achieved hormonal remission in one of the six patients with giant GH-secreting PA. A recent study investigated the effects of switching to pasireotide in a group of acromegalic patients without adequate control under octreotide. Biochemical control was reached in 17.3% of patients treated with pasireotide and none of those remaining on octreotide therapy. 54.3% of pasireotide LAR and 42.3% of octreotide LAR patients achieved significant (≥20%) tumor volume reduction. The safety profile of pasireotide LAR was similar to that of octreotide LAR, with the exception of the frequency and degree of hyperglycemia-related adverse events.98
DA have found their therapeutic place in patients of non-aggressive acromegaly and with little secretory activity. Nevertheless, it has been reported that these compounds may improve the response rate to SSA, and combination therapy with SSA and pegvisomant may be an option in aggressive and non-responder patients with acromegaly. Recent studies demonstrate IGF-1 normalization in 30–40% and 12.5mm3 reduction in tumor volume in octreotide-resistant patients treated with cabergoline and octreotide.99–101
Pegvisomant, a GH receptor antagonist, has shown its efficacy in ameliorating hormonal control in patients with aggressive somatotropinomas resistant to standard therapy with SSA. Percentages of IGF-1 normalization under combination of pegvisomant with SSA have been variable, ranging from 57 and 97%.99,102,103 Rates of tumor growth under pegvisomant therapy have been reported to be 2.9–5.3%.104–106 Some risk factors for this tumor growth have been recently recognized, such as the absence of previous radiotherapy, short duration of previous treatment with SSA, elevated baseline levels of GH, higher increase in GH during treatment with pegvisomant, and higher tumor expression of GH and insulin receptor.104–107 The exon 3 deletion in the GH receptor (GHR) predicts an improved response to pegvisomant therapy in acromegaly according to some authors,108 although this finding has not confirmed in a recent study.109 Pegvisomant may be useful in patients that have insulin resistance and acromegalic cardiomyopathy 110,111; however, pegvisomant would not be indicated as monotherapy in patients with aggressive tumors.
Combination therapy with pegvisomant and SSA may also be useful for patients poorly controlled by conventional approaches. This combination therapy is more likely to be prescribed for patients with biochemical and imaging evidence of aggressive disease.112 In a cohort of 62 acromegalic patients refractory to somatostatin analogs, Bianchi et al.112 showed that there was no significant difference between the daily pegvisomant doses in patients treated with this drug in monotherapy vs. those treated in combination with SSA. However, the final pervisomant dose increased with treatment duration. In the retrospective analysis by Shimon et al. (2015)89 nine patients were treated with pegvisomant reaching remission in 5 of them and partial control (IGF-1<1.5× ULN) in 2 of them. Nevertheless, 5 of these 9 patients were treated with pegvisomant in combination with SSA.
Clinical experience with TMZ in the therapy of aggressive somatotropinomas is limited. In the European Society of Endocrinology survey5 the authors reported the following radiological responses in 14 patients with aggressive somatotroph adenomas: complete regression in 7%, partial regression in 36%, stable disease in 29% and progression in 29%. These values were very similar to those reached in the whole cohort of aggressive pituitary tumors. In this series complete response was only seen in patients with low MGMT expression. Compared to tumors associated with clinical symptoms of acromegaly and elevated serum GH and IGF-1 levels, silent GH adenomas are larger, less differentiated and more aggressive.113–115 Some of them have been reported to be resistant to TMZ.116
Improved responses to TMZ have been reported when this drug is given concurrently with radiotherapy5,6,117 and there are experimental data supporting a radiosensitizing effect of TMZ.118 Nevertheless, progression after TMZ monotherapy has been reported in aggressive somatotropinomas.5,49 Combination treatments with TMZ and capecitabine or TMZ and pasireotide have been used in these cases.6,119 Alternative therapies in TMZ-resistant patients include several chemotherapeutic agents usually in combination. Partial responses have been reported in aggressive somatotroph adenomas with combinations of doxorubicin and CCNU120 and with methotrexate and 5-fluorouracil.121 Targeted therapies (mainly targeting VEGFR and EGFR) are potentially useful in these patients.5,78,122 Some new agents, such as ATL1103, a second-generation, antisense oligomer designed to inhibit translation of human GHR mRNA,123 has been evaluated in a randomized, open-label, phase 2 study in acromegaly, but its potential usefulness in the somatotroph aggressive adenoma must be elucidated.
Among other possible alternative oncological treatment (non-TMZ drugs and radiotherapy) as second and third line treatments are capecitabine, everolimus, and tyrosine kinase inhibitors (TKI).5 Recently, in vitro studies have shown that the RET inhibitor, sorafenib, through AMPK, blocking the GDNF/AKT survival action without altering the RET apoptotic pathway, would be considered as a potential therapeutic alternative in resistant acromegaly.124 A suggested therapeutic approach is shown in Fig. 3.
Aggressive gonadotroph adenomaMost gonadotropic adenomas or gonadotropinomas are silent and manifest clinically related to mass effect. These adenomas are usually slow-growing tumors, behaving as aggressive tumors in a small percentage of cases compared with corticotroph, lactotroph, or somatotroph adenomas.125 In fact, as mentioned above, in large series, gonadotropic adenomas constitute the penultimate histological type of APT.5
Gonadotropinomas show high D2R expression.126 Therefore, the use of DA might have a therapeutic role in aggressive tumors.127
Gonadotroph adenomas also express SST, such as SST2, SST3 and SST5.126,128,129 Among them, SST3 is the most abundant, while sstr5 is expressed in a few percentage of tumors.129 Other studies, however, have shown a higher immunostaining score for SST2 than that for SST3 or SST5 in gonadotroph adenoma and null cell adenoma.128 These findings might be accompanied by therapeutic implications in those tumors that behave more aggressively.128 First-generation SSA, such as octreotide and lanreotide have not been shown to be effective in these tumors, probably due to the low SST2 and SST5 tumor expression. Due to the fact that SST3 expression was high in potentially aggressive lesions, without change in those tumors that recurred after radiotherapy, it seems reasonable to think that the use of a multireceptor ligand SSA like pasireotide can have a therapeutic application in aggressive gonadotrophs.129
The greater expression of D2R than SST would support the medical treatment with DA as first-line therapy in gonadotroph adenomas. The high co-expression of D2R with SST3 in these tumors would support the combined treatment with cabergoline and pasireotide in those more aggressive tumors.
From a series of 10 aggressive non-functioning PAs treated with TMZ, 7 patients had stable disease, 2 patients had reduction of tumor size within 3 months from start of TMZ therapy, and 1 patient tumor had progressive disease.117Fig. 3 shows a therapeutic approach for aggressive gonadotroph adenoma.
Aggressive thyrotroph adenomaMost TSH-secreting PAs express SST.130–132 Hence, SSA have been used as primary treatment or adjuvant to surgery.133,134 In fact, octreotide reduces TSH levels in more than 90% of cases, restores a euthyroid state in the majority of patients and decreases tumor size in nearly half of patients.135
Normalization of thyroid function was achieved in 40 out of 48 patients (83%) treated with SSA in the retrospective study by Yamada et al.136. Tumor shrinkage was found in 24 of 44 patients (55%) treated with these drugs before surgery in this study. Most of patients (82%) in this cohort exhibited macroadenomas, however the Ki-67 labeling index was less than 3% in 97% of tumors for which this marker was available. Therefore, although this series may be representative of TSH-secreting PAs in general, it is likely that it is not for aggressive thyrotroph tumors. Three out of 18 patients with TSH-secreting adenoma retrospectively reviewed by Van Varsseveld et al.137 received SSA therapy exclusively, resulting in apparent cure in one of them. During long-term follow-up, 72% of all patients required medical therapy (mostly SSA treatment), and euthyroidism was achieved in all but one patient, who refused all treatments. These authors conclude that primary medical therapy may be considered in virtually all patients, except in case of optic chiasm compression, especially in those harboring large adenomas with parasellar extension. DA therapy has been employed in some patients with variable results, best responses being obtained in mixed thyrotroph-lactotroph adenomas.138
TMZ has been employed very infrequently so far in aggressive TSH-secreting PAs, and information is scarce and limited. In the recently reported European survey on aggressive PAs, only 1 out of 4 patients with aggressive thyrotroph adenoma reached partial regression, whereas 3 patients attained stable disease after TMZ monotherapy.5 Cytotoxic drugs in combination have been employed in isolated cases of TSH secreting carcinomas.139 A suggested therapeutic approach for aggressive thyrotroph adenoma is shown in Fig. 3.
ReinterventionGoal of surgery should be the maximum possible safe resection, focusing on neural decompression (optic and oculomotor nerves) but without taking excessive risks looking for radical resections. Avoiding surgical complications is mandatory in order to prevent delaying complementary treatments that these patients will normally require. Although total resections in these aggressive tumors is rarely achieved, surgery plays a fundamental role in first-line treatment, and is probably the best treatment for recurrences (at least for the first one).6,10,140 Another advantage of surgery is that it is the only therapy that allows to obtain tissue samples for histopathological study; it is important especially taking into account that tumors can vary their histological characteristics of aggressiveness over time.6,141,142 It is also the most effective and fastest way to decompress optic pathways, and oculomotor nerves in case of cavernous sinus invasion.143,144 It reduces tumor mass decreasing the target volume of radiotherapy procedures, increasing the tumor-quiasm space, making the radio-surgical treatment safer.145 Moreover, it could have some beneficial effect by improving the susceptibility to medical treatments.146,147
Surgical treatment of these lesions is a real challenge that should be performed in reference centers by experienced surgical teams.6 These tumors are usually huge and invasive, with poorly defined borders affecting several paraselar compartments (cavernous sinus, suprasellar area, clivus, sphenoid sinus, etc.). In addition, loss of anatomical references by scar tissue (usually hard and fibrous) due to previous interventions is characteristic. Therefore, these complex surgeries are associated with a lower rate of complete resection and higher morbidity. The postoperative complication rate is higher in comparison with smaller or previously untreated tumors.144,148–153 The most frequent are hypopituitarism, cerebrospinal fluid (CSF) leaks, diabetes insipidus, and nerve structure lesions.
The endoscopic endonasal approach (EEA) is the preferred route in most cases in reference centers, leaving transcranial routes (pterional, subfrontal, andorbitozigomatic) in cases of predominant parasellar or intra-arachnoidal extension. It is also associated with greater comfort and faster postoperative healing compared to the other routes (transcranial and microsurgical transsphenoidal surgery), which allows to perform the subsequent treatments quickly.143 In this type of reoperations, the neuronavigation systems and intraoperative Doppler devices can be very useful to identify intraoperative surgical landmarks.154–156
The main predictor of resecability in PAs is cavernous sinus invasion which can be well systematized by Knosp classification.157 A correlation between the degree of invasion of the cavernous sinus (Knosp grades 3 and 4) and subtotal extirpation has been reported. In fact, most of the tumor remnants in postoperative imaging tests are found in the cavernous sinus.151 Other factors that have been related to subtotal resection are multilobulated tumors, hard-fibrous tumors and those previously treated (operated or irradiated tumors).148,158
There are no reported series regarding surgical outcomes in invasive or aggressive tumors although there are multiple articles regarding surgical resection in giant adenomas or reoperations (both characteristics coexist in these aggressive tumors).148,150,151,153,158,159
Radiation and re-irradiation therapyRadiotherapy (RT) is an essential part of the management of PA with an excellent long-term local tumor control. New techniques include stereotactic radiosurgery (RS), fractionated stereotactic radiotherapy (FSRT), intensity modulated radiotherapy (IMRT) and imaged guided radiotherapy (IGRT). These techniques allow the delivering of higher radiation doses to the target with rapid dose fall-off in the surrounding normal tissues, and potentially limiting the long term toxicity of radiation.160–162
RT is indicated in patients with relevant tumor growth despite surgery in non-functioning or functioning PA that do not respond or do not tolerate medical treatment. When the residual tumor is small, without features of atypia, observation with serial neuroimaging studies are recommended and the RT could be delayed. However, immediate RT is advisable after subtotal surgery for patients with clinically aggressive tumors.163,164
When the standard treatment fails (including RT) several interventions are used before proposing a new irradiation due to morbidity. However some studies of re-irradiation with conventional RT have been published with good results and moderate morbidity.165,166 With the current technical possibilities, a second course of radiation is more feasible with a lower risk of complications.167,168 The choice of technique, always highly conformed, could be RS if the tumor is small, well defined and is not in contact with the optic pathway (Fig. 4). In case of large, invasive tumors close to the optic pathway or difficult to define, FSRT and IMRT are excellent options. The doses administered are usually lower than in a first treatment.169
 of re-irradiation with radiosurgery (Novalis®), dose 12Gy, in a patient with an uncontrollable acromegaly with medical therapy. He had received fractionated stereotactic radiotherapy (54Gy) 12 years before.")
In 2003, Swords et al.,169 analyzed 20 patients, most of them functioning PA (13 acromegaly) treated with linear accelerator-based RS, with a median dose of 10Gy. All patients had received conventional radiotherapy with doses from 45 to 50Gy. They report a rapid decrease in GH and IGF-I levels in all patients with acromegaly, with 50% cure (median follow-up 25 months post radiosurgery) without serious late side effects. Six years later, the same group,170 studied 25 patients, 17 functioning adenomas, treated with RS (gamma knife). They report normalization in IGF-1 levels in 80% of acromegaly patients; with a mean GH level of 1.8ng/ml in 30%. A total of 75% of non-functioning tumors showed disease stabilization or tumor shrinkage. The results were similar with both techniques.
Verma et al.,171 reported 15 patients in which initial RT was delivered using different techniques and re-irradiation was also performed with different modalities. The median dose of re-irradiation was 45Gy for fractionated RT and 18Gy for RS. Optic neuropathy was observed in 13.3%, and temporal lobe necrosis occurred in two patients, both in the group receiving RS. Actuarial local control rates at 2 and 5 years were 80% and 58%, respectively. Four patients (27%) ultimately developed pituitary carcinoma. Re-irradiation is a feasible treatment option in selected cases for local control and hormonal hypersecretion (acromegaly).167 Moderate dose seem to get good results with fewer side effects.
To increase the degree of clinical response, TMZ may be used concomitantly with external beam radiation therapy, as in the Stupp protocol for glioblastoma patients.172 Concurrently with radiotherapy, TMZ is administered daily, including on non radiotherapy weekend days, at a dose of 75mg/m2. In Fig. 5 we present our experience in a non-functioning adenoma with tumor shrinkage. Although the experience in pituitary tumors is anecdotal, the results have been positive.5,173–175 The concomitant therapy with TMZ and radiotherapy has been recommended in the recently reported European Society of Endocrinology Clinical Practice Guidelines for the management of APT and carcinomas, in those patients with rapid tumor growth in whom maximal doses of radiotherapy have not been reached.6
 Re-irradiation in a patient with non-functioning adenoma not controlled neither with surgery nor with RT (previously he had received IMRT 50Gy). Dose distribution with FSRT and IMRT (46.8Gy in 26 fractions). The patient received concomitant therapy with RT and TMZ 75mg/m2 and subsequently 6 cycles of adjuvant TMZ. (B) Pituitary MRI appearances (T1 postgadolinium weighted coronal images). Before (left) treatment and after (right) concurrent therapy with TMZ and radiotherapy. Early response is appreciated.")
(A) Re-irradiation in a patient with non-functioning adenoma not controlled neither with surgery nor with RT (previously he had received IMRT 50Gy). Dose distribution with FSRT and IMRT (46.8Gy in 26 fractions). The patient received concomitant therapy with RT and TMZ 75mg/m2 and subsequently 6 cycles of adjuvant TMZ. (B) Pituitary MRI appearances (T1 postgadolinium weighted coronal images). Before (left) treatment and after (right) concurrent therapy with TMZ and radiotherapy. Early response is appreciated.
The expression of high number of SST2 is the molecular basis for diagnosis and therapy (theragnosis) with somatostatin analogs. SST expression has been demonstrated in all subtypes of pituitary adenoma. Tumoral SST2 can be imaged with octreotide scintigraphy SPECT/TC (Octreoscan®, Tektrotyd®) or 68Ga-DOTATATE positron emission tomography (PET)/CT. Studies evaluating normal tissue uptake of 68Ga-DOTA-TATE/TOC PET/CT report that the normal pituitary gland shows high uptake, although with different SUV values, and no SUV cut-off can differentiate normal pituitary from adenoma.
Peptide receptor radionuclide therapy (PRRT) with radiolabeled somatostatin analogs has been shown to be an effective treatment in metastasized neuroendocrine tumors (NET). Patients with aggressive pituitary tumor with high pituitary radiolabeled SSA uptake, the PRRT (90Y-DOTATOC/TATE and 177Lu-DOTATATE/TOC) is a promising alternative. The efficacy of PRRT in treatment of aggressive pituitary tumors has been demonstrated by single cases or small series published in the last years.
Kaminski et al., 176 reported the first 90Y-DOTATATE treatment in 4 patients with inoperable pituitary tumor. They described partial biochemical response with a decrease of adrenocorticotropic hormone (ACTH) in patients with Nelson's syndrome and a GH decrease in acromegalic patients, and clinical improvement in all cases. Pituitary tumor size was not evaluated.177
Baldari et al.178 reported a significant clinical improvement without side effects after the administration of 4 courses of indium-DTPA-pentetreotide in a patient with recurrent giant prolactin secreting adenoma resistant to standard medical therapy. In 2014 Komor et al.,179 reported a symptomatic improvement, with long-term control in a patient with an atypical non-functioning pituitary adenoma treated with 177Lu DOTATOC. In 2014, Maclean et al. reported not so good results in a 3 patients treated with 177Lu DOTATOC, with only one patient with a clinically evident response.3 In 2015, an acromegalic patient because an invasive macroadenoma treated with of 90Y-DOTATATE achieved partial biochemical remission and a reduction in the tumor size.177
Priola et al.3 selected 7 patients with aggressive pituitary tumor. Three of them with intense pituitary mass uptake in 111In-DTPA-octreotide scintigraphy received PRRT. One patient underwent 5 cycles with 111In-DTPA-octreotide, with a marked tumor size reduction and symptomatic improvement. The other 2 patients showed tumor progression after PRRT.
Maclean et al.,180 observed that patients with rapidly progressive disease with elevated proliferation indices obtained no benefits of treatment with 177Lu-DOTATATE. Secondary local blood flow changes to previous therapy and tumor hypoxia may limit the effectiveness of radiation-based treatments.3
Although the theoretical rationale for PRRT in advanced pituitary adenomas is very attractive, prospective studies are needed to determine patients’ selection, absorbed doses, toxicity and efficacy.
ConclusionsIn recent years, APTs have become a complex clinical challenge and also a gripping area of research for all those interested in pituitary disease. Fortunately, the definition of these pituitary neoplasms has been outlined with some precision in current publications that have studied in depth the behavior of these rare tumors. It is of the utmost importance not to confuse aggressive pituitary tumors with invasive tumors or those that are resistant to the current first line of treatment. Invasiveness and high cell proliferation are characteristic of these tumors, but not enough for their definition.
Precision medicine and personalized medicine are concepts that have recently been introduced in the clinical setting and that fit perfectly into the management of patients with the pituitary neoplasms herein discussed. The approach of these complex patients requires not only a good clinical, analytical, histological and molecular evaluation, but also the case discussion by multidisciplinary teams formed by specialists with expertise in various disciplines and with a work habit that implies joint decision making. Is the opinion of the authors that the multimodal strategy of these patients is a necessity beyond doubt, both in the processes of diagnosis and characterization of tumors (experts in radiology, endocrinology, pathology, genetics, molecular biology) and in the choice of treatments and patient follow-up (specialists in neurosurgery, endocrinology, radiotherapy, oncology and nuclear medicine).
Notwithstanding, gaps in knowledge and barriers still persist which keep us off an adequate approach to these patients in clinical practice. On the other side, the lack of adequate means to know the genetic signatures and the phenotypic expression of receptors and other proteins in each particular patient is common in most health centers dedicated to clinical practice. The centralization of these patients in centers with appropriate expertise and resources should be taken into account by the health authorities to improve the resource performance, increase the quality of health care offered to patients and improve the prognosis of these serious tumors.
