







Non-alcoholic fatty liver disease (NAFLD), a condition that leads to fibrosis, is caused by intake of very high-fat diets (HFDs). However, while the negative impact on the liver of these diets has been an issue of interest, systematic research on the effect of HFDs are lacking.
ObjectiveTo characterize the overall impact of HFDs on both molecular and morphological signs of liver remodeling.
MethodsA study was conducted on male C57BL/6J mice to assess the effect of 4- and 8-week HFDs (60% kcal from fat) on (i) liver steatosis and fibrosis, and (ii) expression of factors involved in inflammation and angiogenesis.
ResultsAfter an 8-week HFD, vascular endothelial growth factor type-2 receptor (VEGF-R2) and fatty acid translocase/trombospondin-1 receptor (CD36) were overexpressed in liver tissue of mice given HFDs. These changes suggest impaired liver angiogenesis and occurred together with (i) increased GPR78-BiP and EIF2α phosphorylation, suggesting endoplasmic reticulum stress, (ii) induction of Col1a1 gene expression, a marker of fibrosis, and (iii) increased CD31 immunolabeling, consistent with active angiogenesis and fibrosis.
ConclusionOur data show that very HFDs promote a rapid inflammatory response, as well as deregulation of angiogenesis, both consistent with development of liver fibrosis.
El hígado graso no alcohólico (HGNA) es una enfermedad hepática que ocasiona fibrosis y se genera por la ingesta de dietas ricas en grasa. Aunque los efectos nocivos de este tipo de dietas son de gran interés, no son muy abundantes los estudios sistemáticos sobre las consecuencias que su consumo puede tener en el hígado.
ObjetivoEvaluar los efectos de una dieta rica en grasa sobre el remodelado hepático, tanto a nivel morfológico como molecular.
MétodosSe utilizaron ratones macho C57BL/6J tratados durante 4/8 semanas con una dieta que contenía un 60% de las kilocalorías procedentes de grasa sobre: 1) la aparición de esteatosis y/o fibrosis hepática y 2) la expresión de factores implicados en procesos de inflamación y angiogénesis.
ResultadosTras 8 semanas de dieta se observó un incremento en el receptor del factor de crecimiento vascular endotelial tipo 2 (R2-FCVE) y en la translocasa de ácidos grasos (CD36). Estos cambios, que sugieren que los procesos angiogénicos del hígado están alterados, fueron simultáneos a: 1) un aumento del GPR78-BiP y de la fosforilación de EIF2α, marcadores de estrés del retículo endoplásmico, 2) la inducción en la expresión del gen de colágeno Col1a1, indicador de fibrosis y 3) un incremento de células inmunofluorescentes para CD31 compatible con procesos de angiogénesis y de fibrosis.
ConclusionesNuestros resultados muestran que las dietas con alto contenido en grasa inducen la rápida activación de respuestas inflamatorias, así como alteraciones en la angiogénesis, siendo ambos procesos compatibles con el desarrollo de fibrosis hepática.
The prevalence of obesity continues to rise in both developing and developed countries and occurs with increasing frequency at early ages. Obesity is a condition associated with a number of health problems that conform the so-called metabolic syndrome, which includes insulin resistance as well as cardiovascular and liver diseases.1 Although the origin of obesity is multifactorial, regular consumption of highly energy-dense foods, rich in saturated fat and sugar, are a main triggering factor.2
Non-alcoholic fatty liver disease (NAFLD) is a common condition in obese people that includes a wide spectrum of liver damage that might progress to liver fibrosis, cirrhosis and hepatocellular carcinoma.3 The pathogenesis of fatty liver is complex and it has been suggested that insulin and leptin resistance are necessary conditions for the accumulation of hepatocellular fat.3,4 In addition, hepatic fat accumulation impairs hepatic insulin sensitivity and promotes steatoinflammatory reactions.5,6 NAFLD can be the consequence of aging and be independent of obesity/overweight,7 but can also be promoted by regular intake of high-fat diets (HFD).4,6 Particular interest has the effect of HFD on hepatic metabolism and function when consumed during the adolescence and early adulthood. As a matter of fact, NAFLD is not only a leading cause of hepatic disease in children but also a relevant factor of cardiovascular risk in this population.8 In addition, it can be assumed that adolescent/juvenile fatty liver is a factor contributing to premature liver aging.7,8
Studies based on experimental models of HFD-induced obesity (DIO) have yielded relevant data illustrating the link between HFD and NAFLD.6 Nevertheless, systematic studies aimed at identifying the effect of HFD during early ages are not abundant in the literature. Moreover, as steatosis can rapidly turn into fibrosis we wonder if this process could be accelerated in young individuals consuming diets containing elevated amounts of saturated fat, as would be the case of fast food diets.6
Because childhood and adolescent obesity has become a major challenge for public health in westernized societies and the prevalence of NAFLD has increased in these populations, our aim was to study the hepatic effects of HFD consumed during the juvenile period in terms of (i) lipid accumulation, (ii) expression of angiogenic/antiangiogenic factors, and (iii) levels of proteins indicative of endoplasmic reticulum stress (ERS), compatible with liver damage.
Material and methodsExperimental designForty 4-week-old male C57BL/6J mice (Harlan, Spain) were housed under a 12-h light/12-h dark cycle in a temperature-controlled room (22°C) with food and water ad libitum. Lights were switched on at 08:00. The study conforms to the National Institute of Health (NIH publication No. 85-23, revised 1996) and was approved by the Ethics Committee of the Universidad CEU-San Pablo. Animals with similar average body weight (BW) were housed, 5 per cage, and assigned either to a high-fat diet (HFD; Test Diet Limited BCM IPS Ltd., London, UK; D12492; 34.9% fat – 14.3% saturated fatty acids, 20.6% unsaturated fatty acids –, 25.9% carbohydrates – 10.55% maltodextrin, 8.85% sucrose and 6.5% powdered cellulose –, 23.1% protein, 6.5% fiber, 9.6% minerals and vitamins; energy density 5.10kcal/g; n=20) or to a standard rodent chow (SD; Teklad, Harlan, Spain; 6.2% fat – 1.1% saturated fatty acids, 5.1% unsaturated fatty acids –, 44.2% carbohydrates – wheat, corn, wheat midds and corn gluten meal–, 18.6% protein, 14.7% fiber, 16.3% minerals and vitamins; energy density 3.1kcal/g; n=20). BW and food intake were monitored once a week. HFD mice and their respective control littermates had free access to food either during 4 or 8 weeks. On the last day of treatment, free-feeding mice were weighed and killed by decapitation between 09:00 and 11:00. Blood was collected in chilled EDTA-coated polypropylene tubes, and tissues dissected, weighed, and either frozen in liquid nitrogen or fixed in formalin.
Plasma biochemistry, insulin resistance assessment (HOMA-IR) and glucose tolerance test (GTT)Plasma leptin and insulin concentrations were analyzed by using specific ELISA kits provided by Phoenix Europe GmbH (Germany) and Mercodia (Spain), respectively. Glucose (Glucose Trinder Method; Roche, Spain), triglycerides (glycerol-phosphate oxidase method; Biolabo, France), and non-esterified free fatty acids (NEFA) (acyl-CoA synthase-acyl-CoA oxidase method, Wako Bioproducts, Richmond, VA) were measured by colorimetric methods. Homeostasis model assessment for insulin-resistant (HOMA-IR) indexes were calculated, after 6h fasting, 5 days before animals were killed, as previously reported.9 For GTT assessment mice were fasted 6h before glucose loading (i.p.; 1g/kg). Blood glucose levels were measured (0, 30, 60, 90 and 120min after injection) by means of an Accu-Check Aviva glucometer (Roche Diagnostics, Mannheim, Germany).
Measurement of total lipid content in liverHepatic lipids were extracted in chloroform/methanol (2/1) following the method of Folch with modifications.10
Western blottingLiver samples were homogenized in ice-cold buffer containing 0.42M NaCl, 20mM HEPES (pH 7.9), 1mM Na4P2O7, 1mM EDTA, 1mM EGTA, 1mM dithiothreitol, 20% glycerol, 1mg/ml aprotinin, 1mg/ml leupeptin, 20mM sodium fluoride, 1mM trisodiumorthovanadate, and 2mM phenylmethylsulfonyl fluoride. The homogenates were frozen at 80°C and thawed at 37°C three times and then centrifuged for 10min at 4°C. Equivalent amounts of protein (30μg) were loaded in Laemmli buffer (50mM Tris pH=6.8, 10% sodium dodecyl sulfate, 10% glycerol, 5% mercaptoethanol, and 2mg/ml blue bromophenol) and size separated by SDS-PAGE. Samples from plasma (7μg) were loaded in Laemli without previous manipulation. In both cases, proteins were transferred to polyvinylidenedifluoride membranes (GE Healthcare, Barcelona, Spain) by using a transblot apparatus (Bio-Rad, Hercules, CA). The membranes were blocked with 5% nonfat dried milk in Tween-PBS for 1h. Primary antibodies against ANGPTL-4 (dilution1:1000; anti-ANGPTL-4 antiserum was directed against the 80-94 residues, located in the N-terminal domain of the protein, Abcam, Cambridge, UK), p-EIF2α (dilution 1:1000; Abcam, Cambridge, UK) and GPR78-BiP (dilution 1:1000; Cell Signalling, USA) were applied at the convenient dilution overnight at 4°C. After washing, appropriate secondary antibodies (anti-rabbit IgG-peroxidase conjugated) were applied for 1h at a dilution of 1/5000. Blots were washed, incubated in enhanced chemoluminescence reagent (ECL; GE Healthcare). Films were scanned using a GS-800 calibrated densitometer (Bio-Rad) and blots were quantified by using QuantityOne software (Bio-Rad). Values for GPR78-BiP were normalized with β-actin (Affinity Bioreagents, Golden, CO). Phosphorylated EIF2α (p-EIF2α) was normalized with EIF2α (Abcam, Cambridge, UK). Plasma ANGPTL-4 values were normalized with serum albumin that was quantified in membranes after staining with Ponceau S red (Sigma–Aldrich, Spain).
Hepatic lipase activityFifty mg samples were homogenized in 0.8ml 0.2M Tris–HCl buffer (pH=8.2). After centrifugation (10min, 3000rpm), the aqueous phase was transferred to eppendorf tubes containing 50μl preheated fetal calf serum (60°C; 10min). Proteins were precipitated with cold acetone (−20°C), then washed with diethyl ether and gently dried under a helium stream. The resulting powder was re-suspended in 1ml 0.05M NH4OH/NH4Cl buffer (pH=8.1). After protein quantification, samples were assayed for hepatic lipase activity as described.11 Briefly, 0.2ml samples were incubated with 0.05ml distilled water and 0.1ml reaction mixture prepared from (i) 0.04ml 0.2M Tris–HCl buffer (pH=8.2), containing 6% fatty acid free BSA and 1M NaCl as inhibitor of other lipases, (ii) 0.02ml preheated fetal calf serum, and (iii) 0.04ml C14-triolein solution (69mg triolein, 3.3mg lecithin, 462kBq C14-triolein, and 5ml glycerol). C14-triolein was purchased from Perkin Elmer (Spain). Plasma from rats receiving 12UI heparin/100g BW (15min before bleeding) was used as a positive control. After 30min incubation (37°C) the enzymatic reaction was stopped by the addition of 2ml chloroform/heptane/methanol (1/1/1), and free C14-oleic acid extracted with 1ml 0.1M KBO3/K2CO3 (pH=8.5). After centrifugation (15min; 3000rpm), the radioactivity contained in 1ml of the upper aqueous phase was quantified in a scintillation counter. All samples were run in triplicate. Enzymatic activity was measured both after 4- and 8-week dietary treatment and was expressed in nmol/min/mg protein.
RNA preparation and quantitative real-time PCRTotal RNA was extracted from liver by using the Tri-Reagent protocol (Sigma Chemical Co., St. Louis, MO). cDNA was then synthesized from 2μg total RNA by using a high-capacity cDNA RT kit (Applied Biosystems, Foster City, CA). Quantitative RT-PCR was performed by using an assay-on-demand kit (Bio-Rad) for fatty acid translocase CD36 (Cd36, Mn01135198_m1), and designed primer pairs (Integrated DNA Technologies, USA) for vascular endothelial growth factor type-2 receptor (Vegf-R2, forward 5′-CCTGAGAACTGGGCTCTGTG-3′, reverse 3′-ATGGAGAAAATCGCCAGGCA-5′), interleukin 6 (Il-6, forward 5′-AGCCAGAGTCCTTCAGAGAGA-3′, reverse 5′-GGAGAGCATTGGAAATTGGGG-3′) and collagen 1 (Col1a1, forward 5′-ACTGCCCTCCTGACGCAT-3′, reverse 5′-AGAAAGCACAGCACTCGCC-3′). TaqMan Universal PCR Master Mix (Applied Biosystems, USA) and SsoAdvanced Universal SYBR Green Supermix (Biorad, Spain) were used, respectively, for amplification according to the manufacturer's protocols, in an ABI PRISM 7000 Sequence Detection System (Applied Biosystems, USA). Values were normalized to the housekeeping gene β-actin (Actb Mn00607939 s1) and 18S ribosomal RNA (18S, forward 5′-GGGAGCCTGAGAAACGGC-3′, reverse 5′-GGGTCGGGAGTGGGTAATTT-3′). According to manufacturer's guidelines, the ΔΔC(T) method was used to determine relative expression levels. Statistics were performed using ΔΔC(T) values.12
Hematoxylin/eosin and Sirius red stainingTissue samples were fixed in 4% formaldehyde for 10 days, then washed in water (2h), dehydrated with ethanol and subsequently embedded in paraffin. Thin serial sections (5μm) were obtained with a vertical rotary microtome (Leica RM 2125RT), and mounted in glass slides. Slices were stained with (i) hematoxylin/eosin (H/E; Thermo Scientific, Spain), for parenchyma evaluation and cellular morphology, and (ii) Sirius red (Sigma, Spain) for collagen. After staining, sections were dehydrated in ethanol and xylene and mounted with DPX. Samples were directly observed (20×) by using an Eclipse 50i-Nikon microscope equipped with a camera (DS-5M) and a Nis-Elements software.
Immunofluorescence staining of cluster of differentiation 31 protein (PECAM-1/CD31)Tissue sections (10μm) were obtained on a Criocut (Leica CM 1850), then bathed sequentially in PBS/0.5% Triton (20min), PBS/3% BSA (30min) and finally incubated overnight (4°C) in immunofluorescence (IF) buffer containing PBS, 0.1% BSA, 0.2% Triton and 0.05% Tween with primary anti-CD31 (dilution 1:50, Santa Cruz Biotechnology, USA). After washing in IF buffer, samples were incubated for 1h with a secondary antibody against CD31 (anti-goat, dilution 1:100, Alexa 647, Abcam). Nuclei were stained with DAPI (dilution 1:5000, Invitrogen, Spain).13 Fluorescence analysis was performed with a Leica TCS SP5 confocal microscope and relative intensity was quantified with the ImageJ software.
StatisticsIndividual group comparisons in the assay carried-out in mice were made using a one-way ANOVA, followed by Newman–Keuls’ post hoc test. Statistical significance was set at p<0.05. All statistics were performed by using GraphPad Prism 7 (GraphPad Sofwares, USA).
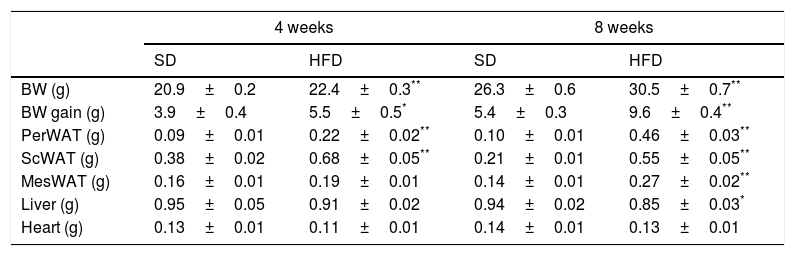
ResultsEffect of HFD on adiposity and body weightTable 1 summarizes the effect of dietary treatments on liver, heart and body weights as well as in the amount of visceral (perirenal – PerWAT–; mesenteric – MesWAT) and subcutaneous (ScWAT) white adipose tissues. Four-week of HFD increased BW (p<0.001), PerWAT (p<0.001) and ScWAT (p<0.001), an effect that was extended to the MesWAT after 8-week dietary treatment (p<0.001). In contrast, liver weight exhibited a modest decrease after 8-week of HFD (p<0.05). No effect was detected on heart weight.
Effect of four- and eight-week HFD intervention on BW and weight of perirenal (PerWAT), subcutaneous (ScWAT) and mesenteric (MesWAT) adipose tissue, liver and heart.
| 4 weeks | 8 weeks | |||
|---|---|---|---|---|
| SD | HFD | SD | HFD | |
| BW (g) | 20.9±0.2 | 22.4±0.3** | 26.3±0.6 | 30.5±0.7** |
| BW gain (g) | 3.9±0.4 | 5.5±0.5* | 5.4±0.3 | 9.6±0.4** |
| PerWAT (g) | 0.09±0.01 | 0.22±0.02** | 0.10±0.01 | 0.46±0.03** |
| ScWAT (g) | 0.38±0.02 | 0.68±0.05** | 0.21±0.01 | 0.55±0.05** |
| MesWAT (g) | 0.16±0.01 | 0.19±0.01 | 0.14±0.01 | 0.27±0.02** |
| Liver (g) | 0.95±0.05 | 0.91±0.02 | 0.94±0.02 | 0.85±0.03* |
| Heart (g) | 0.13±0.01 | 0.11±0.01 | 0.14±0.01 | 0.13±0.01 |
Values are means±S.E.M. of 10 individual values.
As illustrated in Fig. 1, HOMA-IR indexes (Fig. 1A), estimated from glucose and insulin concentration after 6h of fasting (Fig. 1B and C, respectively), revealed an impaired sensitivity to insulin in DIO mice after an 8-week HFD (p<0.01). In this cohort, plasma NEFA were also increased (p<0.05; Fig. 1D). Plasma TGs were not modified by HFD (Fig. 1E), while plasma leptin (Fig. 1F) appeared to be increased in HFD mice both after 4- (p<0.01) and 8-week treatments (p<0.001). GTT revealed an impaired management of glucose after both 4- and 8-week HFD (p<0.01; Fig. 1G and p<0.001; Fig. 1H).
. Plasma glucose after 6h fasting was increased after 8w-diet (B) but insulin concentration was not modified (C). HFD increased plasma NEFA only after 8w-diet (D) but plasma TG were not modified (E). Plasma leptin (F) was increased after both 4- and 8-week of dietary treatment. Glucose tolerance was impaired both after 4- (G) and 8-week HFD (H). Graph bars represent means±S.E.M. of 10 values (*p<0.05, **p<0.01, ***p<0.001, compared to control groups; Newman–Keul")
Effect of HFD on food intake, plasma parameters and glucose tolerance. HOMA indexes were only altered after 8-week HFD (A). Plasma glucose after 6h fasting was increased after 8w-diet (B) but insulin concentration was not modified (C). HFD increased plasma NEFA only after 8w-diet (D) but plasma TG were not modified (E). Plasma leptin (F) was increased after both 4- and 8-week of dietary treatment. Glucose tolerance was impaired both after 4- (G) and 8-week HFD (H). Graph bars represent means±S.E.M. of 10 values (*p<0.05, **p<0.01, ***p<0.001, compared to control groups; Newman–Keul's test).
As illustrated in Fig. 2A, HFD enhanced total lipid content after 4-week treatment (p<0.01) and tended to increase it after 8 weeks (p=0.06). Concentration of hepatic lipids appeared to be also increased in HFD mice after both 4- (p<0.05) and 8-week dietary treatment (p<0.001; Fig. 2B). In order to evaluate the influence of HFD on general morphology as well as on the eventual accumulation of intrahepatic lipids and fibrosis, liver sections were stained with either H/E (Figs. 2C–F) or Sirius red (Fig. 2G–J). As illustrated in panels Fig. 2C–F, H/E staining was compatible with hepatic steatosis in mice that consumed the HFD. Vacuolization of hepatocytes was detected both after 4-week (Fig. 2D) and 8-week (Fig. 2F). Fig. 2G–J shows representative micrographs of Sirius red staining, revealing fibrillar collagen deposition in samples from 4- and 8-week HFD mice. Coherently, the expression of the collagen gene Col1a1 appeared to be induced both after 4- (p<0.001) and 8-week HFD (p<0.01) (Fig. 2K).
 and relative (B) hepatic lipids were increased after 4- and 8-week HFD (B). Photomicrographs show representative images (x20) of H/E (C–F) and Sirius red staining (G–J). Lipid vacuoles (black arrows) were observed in photomicrographs (D) (4-week HFD) and (F) (8-week HFD). Collagen deposition, indicative of perivascular fibrosis, was well defined in samples from 4- (H) and 8-week HFD mice (J). HFD promoted a significant increase of Col1a1 expression after 4- and 8w-diet (K). Col1a1 value was normalized to the housekeeping gene β-actin and 18S ribosomal RNA. Graph bars represent means±S.E.M. of 10 values (*p<0.05, **p<0.01, ***p<0.001, compared to control groups; Newman–Keul")
Effect of HFD on hepatic lipids and liver morphology. Both total (A) and relative (B) hepatic lipids were increased after 4- and 8-week HFD (B). Photomicrographs show representative images (x20) of H/E (C–F) and Sirius red staining (G–J). Lipid vacuoles (black arrows) were observed in photomicrographs (D) (4-week HFD) and (F) (8-week HFD). Collagen deposition, indicative of perivascular fibrosis, was well defined in samples from 4- (H) and 8-week HFD mice (J). HFD promoted a significant increase of Col1a1 expression after 4- and 8w-diet (K). Col1a1 value was normalized to the housekeeping gene β-actin and 18S ribosomal RNA. Graph bars represent means±S.E.M. of 10 values (*p<0.05, **p<0.01, ***p<0.001, compared to control groups; Newman–Keul's test).
As inflammation and angiogenesis are closely related entities, we analyzed the influence of HFD on the expression of some factors/receptors endowed with either proangiogenic (VEGF-R2, type-2 receptor of vascular endothelial growth factor) or antiangiogenic activity (CD36, ANGPTL-4), that could also participate in inflammatory processes.
An up-regulation of Vegf-R2 expression was detected both after 4- and 8-week HFD (p<0.05; Fig. 3A) and was coherent with a concomitant increase of PECAM-1/CD31 immunoreactivity defining a dense network of endothelial cells that was less intense in controls than in HFD mice after 8-week dietary intervention (Fig. 3B–F, p<0.05). As illustrated in Fig. 3G, circulating ANGPTL-4 immunoreactivity was increased by HFD after 4-week HFD (p<0.05), but was reduced after 8-week treatment (p<0.05). Because ANGPTL-4 is a negative modulator of hepatic lipase,14 the activity of this enzyme was evaluated. We observed an increase of hepatic lipase activity after 8-week of HFD (p<0.05, Fig. 3H), compatible with the decrease of circulating ANGPTL-4.
. Expression of PECAM-1/CD31 was assessed in 8-week HFD liver samples by means of immunofluorescence staining (PECAM-1/CD31, red; DAPI-stained nuclei, blue; 40×) (B–E). Relative intensity of CD31 immunofluorescence is illustrated in panel (F). Four-week HFD evoked an increase of plasma ANGPTL-4 immunoreactivity, while 8-week treatment decreased the amount of this protein (G). Accordingly, hepatic lipase activity was increased in this cohort (H). Cd36 gene expression was also increased in 8-week HFD mice (I). Hepatic GPR78-BiP immunoreactivity was decreased after 4-week HFD but was increased after 8-week HFD (J). Variations of hepatic p-EIF2α/EIF2α were qualitatively similar to those observed in GRP78-BIP, although they did not reach statistical significance (K). Interleukin 6 (Il-6) was up-regulated only after 8-week HFD (L). Vegf-R2, Cd36 and Il6 values were normalized to the housekeeping gene β-actin and 18S ribosomal RNA. Values are means±S.E.M. of 10 individual values. *p<0.05, **p<0.01, ***p<0.001, compared to their correspondent control groups (SD) (Newman–Keul")
Effect of HFD on the expression of angiogenic, antiangiogenic and pro-inflammatory factors. HFD evoked a significant up-regulation of Vegf-R2 expression both after 4- and 8-week HFD (A). Expression of PECAM-1/CD31 was assessed in 8-week HFD liver samples by means of immunofluorescence staining (PECAM-1/CD31, red; DAPI-stained nuclei, blue; 40×) (B–E). Relative intensity of CD31 immunofluorescence is illustrated in panel (F). Four-week HFD evoked an increase of plasma ANGPTL-4 immunoreactivity, while 8-week treatment decreased the amount of this protein (G). Accordingly, hepatic lipase activity was increased in this cohort (H). Cd36 gene expression was also increased in 8-week HFD mice (I). Hepatic GPR78-BiP immunoreactivity was decreased after 4-week HFD but was increased after 8-week HFD (J). Variations of hepatic p-EIF2α/EIF2α were qualitatively similar to those observed in GRP78-BIP, although they did not reach statistical significance (K). Interleukin 6 (Il-6) was up-regulated only after 8-week HFD (L). Vegf-R2, Cd36 and Il6 values were normalized to the housekeeping gene β-actin and 18S ribosomal RNA. Values are means±S.E.M. of 10 individual values. *p<0.05, **p<0.01, ***p<0.001, compared to their correspondent control groups (SD) (Newman–Keul's test).
Fig. 3I shows the effect of HFD on CD36 expression both after 4- and 8-week of dietary treatment (p<0.001). CD36 is a pleiotropic protein, endowed with antiangiogenic and pro-inflammatory properties. Its up-regulation is coherent with a pro-inflammatory state, suggested by the enhanced levels of expression of GPR78-BiP (Fig. 3J; p<0.05) and the phosphorylated form of p-EIF2α (Fig. 3K; p=0.06). In addition, we observed a significant increase of Il-6 mRNA after 8-week HFD (Fig. 3L; p<0.01).
DiscussionOur study shows that 8-week consumption of a diet providing 62% kcal. from lard during the juvenile period triggers liver damage beyond steatosis. These kind of diets have been shown to evoke typical metabolic disorders frequently observed in obese humans,15,16 and they are widely used to model in rodents the metabolic remodeling associated to human obesity. In fact, several studies have evidenced that obese patients present elevated risk of hepatic dysfunction, including NAFLD and hepatocellular.17
To our knowledge this is the first study carried out on adolescent/young mice focused on the effect of HFD on hepatic angiogenesis and inflammation. The most striking result is the rapid evolution from steatosis toward liver inflammation and fibrosis that mimics the natural process of liver aging.7 We have detected the up-regulation of the trombospondin receptor CD36, the endothelial cell marker PECAM-1/CD31, and the type-2 receptor of VEGF, which are all involved in inflammatory processes as well as in driving angiogenesis.18
We first identified an increase in hepatic lipids after 4-week HFD that was perceptible before the impairment of insulin sensitivity was established. In fact, the only apparent endocrine alteration observed at this time was a small raise of plasma leptin, which was associated to the increase of adiposity. The liver also exhibited a normal histology, with the exception of scattered lipid droplets, and signs of fibrosis were absent. This suggests that the increase of hepatic lipids at this stage of the dietary treatment is a physiological adaptive process aimed at managing the overload of dietary lipids. Accordingly, none of the pro-inflammatory markers analyzed in the study were significantly modified. In contrast to that, steatosis detected after 8-week HFD occurred concomitantly with insulin resistance, a dramatic increase of plasma leptin, elevated plasma NEFA in non-fasted individuals, increased expression of Cd36 (300%) and Vegf-R2 as well as an increased phosphorylation/expression of ERS markers within the liver. The histology, in this case, was compatible with a pre-fibrotic/fibrotic state characterized by collagen deposits and a dense expression of CD31 in the hepatic vascular network.19 Overall, the study shows that the capacity of mouse liver to accumulate lipids during a dietary overload is rapidly overcame and progresses toward a pre-pathological/pathological state. This issue is particularly relevant as hepatic fibrosis, which is not necessarily an irreversible process, can promote further hepatic damage and lead to premature liver aging.7,20
One of the most striking results of the current study is the dramatic up-regulation of Cd36 expression (Fig. 3D). CD36 is a class B scavenger receptor involved in angiogenesis, atherosclerosis, inflammation and lipid metabolism,21 whose increase is consistent with previous research demonstrating its contribution to hepatic steatosis during HFD consumption.22 Moreover CD36 is a receptor essential for the action of pro-inflammatory factors, that also contributes to impair insulin signaling and its increase can be considered as a marker of insulin resistance.23 CD36 seems also to have a pivotal role in diet-induced lipotoxicity, as Cd36 gene expression has been shown to promote hepatic lipotoxic damage.22,23 Otherwise, CD36 is a negative regulator of angiogenesis able to counterbalance the proliferative effect of VEGF.24 In our study we have detected a slight up-regulation of the subtype-2 receptor of VEGF (Vegf-R2) (Fig. 3A), a result coherent with the fact that endothelial VEGF-R2 and CD36 can form molecular complex endowed antiangiogenic action.18 Another, still not well-defined, role of CD36 deals with its participation in ERS.25 We have observed that the Cd36 up-regulation occurs concomitantly with an enhanced expression of GRP78-BIP, phosphorylation of EIF2α and Il-6 (Fig. 3G, H and I, respectively), compatible with ERS and coherent with previous studies showing that chronic HFD promotes ERS in hepatocytes.26 Finally, CD36 is also a carrier contributing to FA uptake by hepatocytes. Although the participation of CD36 in this process seems to be quite modest under physiological conditions, some studies point to a more important role in FA uptake during HFD treatment. Moreover, under insulin resistance and elevated levels of plasma NEFA (our data), CD36 might be pivotal to facilitate hepatic FA uptake.27
Another relevant result of the current study is the intense CD31 immunoreactivity observed in liver slices after 8-week HFD, which is coherent with a fibrotic state. CD31 is an endothelial marker whose hepatic expression increases during pathological angiogenesis and concomitantly with the progression of liver fibrosis.28 The regulation of CD31 by HFD has not been, to our knowledge, the object of previous studies, but its up-regulation is coherent with the already reported pro-angiogenic effect of HFD in the hepatic tissue.29 Finally, a role of leptin/hyperleptinemia can be also hypothesized to explain the increase of CD31, as leptin-mediated neovascularization has been shown to be a pre-requisite for the progression of NAFLD in rats.30 Although hepatic steatosis has been related with the development of hepatic leptin resistance rather than with the effect of leptin,4,31 the role of endothelial leptin receptors has not been properly established. In fact, endothelial leptin receptors seem to activate the PI3K/Akt pathway32 rather than the JAK/STAT3 pathway, whose desensitization has been shown to promote steatosis.4 The decrease of circulating ANGPTL-4 triggered by 8-week HFD is coherent with the enhancement of hepatic lipase activity, which might contribute to plasma TG clearance and, therefore, to hepatic steatosis14 and would be compatible with an inflammatory state.33
It is important to note that hypercaloric diets used in this kind of studies are usually rich in sucrose, which has been shown to promote the production of pro-inflammatory mediators and impair insulin signaling.34 Therefore, both fat and sugar may contribute to the development of hepatic alterations such as non-alcoholic steatosis and cirrhosis.35,36
In summary, our study shows that exposure to HFD during the juvenile period promotes a pro-inflammatory state together with a deregulation of angiogenic/antiangiogenic factors. Both molecular and structural alterations triggered by HFD might influence to the development of liver fibrotic and induce premature liver aging, as well. Although our study highlights the deleterious effect of HFD in early ages, the responsible mechanisms remain to be fully elucidated.
FundingThis work was supported by Fundación Universitaria San Pablo-CEU and Ministerio de Economía y Competitividad, Spain (BFU2012-35353; BFU2016-78556-R). A.P. is supported by a grant from Ministerio de Economía y Competitividad (BES-2013-063773). A.M.B. is supported by the European Erasmus Programme (KU Leuven, Belgium).
Conflict of interestThe authors have nothing to disclose.
