






Describir la frecuencia, los aspectos clínicos, bioquímicos y moleculares de la hipercolesterolemia familiar (HF) en sujetos que acuden a una unidad de endocrinología.
MétodosEstudio observacional, descriptivo en el que se evaluaron 3.140 sujetos que fueron atendidos en la Unidad de Endocrinología del Centro Médico Orinoco en Ciudad Bolívar, Venezuela, desde el 7 de enero del 2013 al 9 de diciembre del 2016. Los casos índice fueron seleccionados de acuerdo con los criterios de la Red de Clínicas de Lípidos de Holanda. Se midieron lípidos plasmáticos. El análisis molecular se realizó por medio de secuenciación de ADN de los genes LDLR y APOB.
ResultadosDe los 3.140 sujetos evaluados, 10 (0,32%) tuvieron características clínicas y bioquímicas compatibles con HF. Todos, excepto uno, eran de sexo femenino. Tres pacientes tuvieron antecedente familiar en primer grado de enfermedad coronaria prematura y ninguno antecedente personal de esta patología. Tres pacientes tuvieron obesidad, 3 hipertensión arterial y ninguno tuvo diabetes. Tres pacientes presentaban xantomas tendinosos y solo uno arco corneal. Los valores de c-LDL oscilaron entre 191 y 486mg/dl. Solo 2 recibían tratamiento con estatinas. En 4 pacientes se identificó la causa genética de la HF: 3 de ellos por mutaciones en el gen LDLR y uno por mutación en el exón 26 del gen APOB.
ConclusiónAproximadamente una de cada 300 personas que acuden a consulta en esta unidad de endocrinología presentan HF. Las mutaciones en el gen LDLR son las principales causantes de HF en este grupo de pacientes.
To assess the frequency and the clinical, biochemical, and molecular aspects of familial hypercholesterolemia (FH) in subjects attending an endocrinology unit.
MethodsAn observational, descriptive study evaluating 3,140 subjects attending the endocrinology unit of Centro Médico Orinoco in Ciudad Bolívar, Venezuela, from 7 January 2013 to 9 December 2016. The index cases were selected using the Dutch Lipid Clinic Network criteria. Plasma lipid levels were measured, and a molecular analysis was performed by DNA sequencing of the LDLR and APOB genes.
ResultsTen (0.32%) of the 3,140 study patients had clinical and biochemical characteristics consistent with FH. All but one were female. Three had first-degree relatives with prior premature coronary artery; and none had a personal history of this condition. Three patients were obese; three had high blood pressure; and no one suffered from diabetes. Three patients had a history of tendon xanthomas, and one of corneal arcus. LDL-C levels ranged from 191 to 486mg/dL. Two patients were on statin therapy. The genetic causes of FH were identified in four patients, and were LDLR gene mutations in three of them and an APOB gene mutation in exon 26 in the other.
ConclusionApproximately, one out of every 300 people attending this endocrinology unit in those four years had FH, and LDLR gene mutations were the most prevalent cause.
La hipercolesterolemia familiar (HF) (OMIM 143890) es el trastorno monogénico más frecuentemente asociado con enfermedad coronaria prematura, debido a elevadas concentraciones de colesterol transportado por lipoproteínas de baja densidad (c-LDL)1. Se transmite de forma autosómica dominante y se produce principalmente por mutaciones en el gen del receptor de c-LDL (LDLR), y menos frecuentemente por mutaciones en los genes de la apolipoproteína B (APOB) y de la proproteína convertasa subtilisina/kexina tipo 9 (PCSK9)2,3.
La prevalencia de HF heterocigota es de aproximadamente una de cada 300-500 personas en la población general4. Por su parte, la HF homocigota afecta a un caso por 1.000.000 de personas, aunque es mayor en determinadas regiones o países, presumiblemente debido a una elevada tasa de endogamia5.
Los sujetos con HF heterocigota tienen de 3 a 4 veces mayor riesgo de presentar enfermedad arterial coronaria y tienden a desarrollarla, en promedio, una década antes que la población general3,6. Por tanto, la identificación temprana de sujetos con HF resulta fundamental debido a que el tratamiento precoz puede reducir el riesgo de aterosclerosis prematura7; sin embargo, la mayoría de los pacientes no se encuentran diagnosticados ni tratados8.
En Latinoamérica se desconoce la prevalencia de HF y particularmente en Venezuela no existen estudios previos que hayan evaluado la frecuencia de esta enfermedad ni las características clínicas de los sujetos afectados9. Hasta donde tenemos conocimiento, solo un estudio realizado en Maracaibo, Estado Zulia, evaluó en 65 sujetos con hipercolesterolemia, sin diagnóstico clínico de HF, la presencia de mutaciones en el exón 4 del gen LDLR, evidenciando mutaciones en 5 pacientes10. A pesar de ello, en el país existe desconocimiento de la enfermedad por parte de la comunidad médica en general, lo cual, aunado a la falta de políticas públicas que permitan un adecuado registro nacional de pacientes con HF y la ausencia de centros de referencia especializados en lípidos, limita el diagnóstico y tratamiento oportuno de la enfermedad. Por tanto, el objetivo de este estudio es describir la frecuencia, los aspectos clínicos y bioquímicos de la HF en sujetos que acuden a una unidad de endocrinología en Ciudad Bolívar, Venezuela, así como caracterizar las mutaciones capaces de producir HF en este grupo de pacientes.
MetodologíaDiseño del estudio y sujetosCon base en el objetivo propuesto, se diseñó un estudio observacional y descriptivo, en el cual se evaluaron un total de 3.140 sujetos que fueron atendidos en la Unidad de Endocrinología, Diabetes, Metabolismo y Nutrición del Centro Médico Orinoco en Ciudad Bolívar, Venezuela, desde el 7 de enero del 2013 al 9 de diciembre del 2016. Los casos índice con sospecha clínica y bioquímica de HF fueron seleccionados de acuerdo con los criterios de la Red de Clínicas de Lípidos de Holanda (RCLH) (tabla 1)11. El diagnóstico es de certeza cuando la puntuación es >8 puntos y de probabilidad cuando la puntuación es de 6 a 8 puntos11,12. El diagnóstico clínico de HF incluyó a ambos grupos debido a que es posible detectar mutaciones causantes de HF en un número significativo de sujetos con diagnóstico de probabilidad. En el análisis molecular se incluyeron pacientes que obtuvieron una puntuación ≥6 puntos, como recomiendan la Sociedad Europea de Aterosclerosis8 y el consenso para el diagnóstico y tratamiento de la HF en España12.
Criterios de la Red de Clínicas de Lípidos de Holanda para el diagnóstico de hipercolesterolemia familiar
| Característica | Puntuación |
|---|---|
| Historia familiar | |
| Familiar de primer grado con enfermedad coronaria prematura (hombres ¿55 años y mujeres ¿60 años) y/o familiar de primer grado con niveles de c-LDL >210mg/dl | 1 |
| Familiar de primer grado con xantomas tendinosos y/o arco corneal ¿45 años y/o familiar ¿18 años con c-LDL ≥150mg/dl | 2 |
| Antecedentes personales | |
| Paciente con enfermedad coronaria prematura (hombres ¿55 años y mujeres ¿60 años) | 2 |
| Paciente con enfermedad cerebrovascular o arterial periférica prematura (hombres ¿55 años y mujeres ¿60 años) | 1 |
| Examen físico | |
| Xantomas tendinosos | 6 |
| Arco corneal ¿45 años | 4 |
| Análisis de laboratorio | |
| c-LDL ≥330mg/dl | 8 |
| c-LDL 250-329mg/dl | 5 |
| c-LDL 190-249mg/dl | 3 |
| c-LDL 155-189mg/dl | 1 |
| Análisis genético | |
| Mutación funcional en los genes LDLR, APOB o PCSK9 | 8 |
Además, se excluyeron del estudio las mujeres embarazadas, sujetos con historia de abuso de alcohol, pacientes infectados con el virus de inmunodeficiencia humana (VIH) en tratamiento antirretroviral, sujetos con colestasis o insuficiencia hepática, insuficiencia renal crónica, endocrinopatías como hipotiroidismo no tratado y síndrome de Cushing, pacientes con diagnósticos de otras hiperlipidemias primarias, sujetos con concentraciones de triglicéridos ≥400mg/dl, así como aquellos tratados con andrógenos, ciclosporina, amiodarona, ácido retinoico y esteroides. El estudio fue aprobado por el Comité de Ética de la institución y siguiendo los lineamientos propuestos por la Declaración de Helsinki todos los sujetos dieron su consentimiento por escrito para participar en el mismo.
Variables antropométricas y clínicasEl peso y la talla se obtuvieron con los sujetos en ayunas y vistiendo solo su ropa interior. El índice de masa corporal (IMC) se calculó como el peso dividido entre la talla al cuadrado. Se consideró obesidad cuando el IMC fue ≥30kg/m213. En los sujetos con sospecha de HF se evaluaron los tendones, principalmente el tendón de Aquiles y los tendones extensores de las manos a fin de identificar la presencia o no de xantomas, y se observó la periferia del iris a fin de valorar la existencia de arco corneal.
Variables bioquímicasPara la determinación de los lípidos se tomó una muestra de sangre de la vena antecubital en ayuno no menor de 8h. Las cuantificaciones de colesterol total, triglicéridos y colesterol transportado por lipoproteínas de alta densidad (c-HDL) se hicieron por métodos enzimáticos con un autoanalizador Hitachi 911® y reactivos de la casa comercial Cienvar. El c-LDL se estimó a través de la ecuación de Friedewald, donde c-LDL=colesterol total–[c-HDL+(triglicéridos/5)].
Análisis genéticoPara el análisis, se extrajo ADN de muestras de sangre periférica usando el kit de purificación de ADN genómico Wizard® (Promega, EE.UU.). El diagnóstico genético de HF se realizó en 2 fases: la fase 1 incluyó el estudio de las mutaciones más frecuentes en el gen APOB (fragmentos de los exones 26 y 29) y el estudio molecular del promotor, empalme (splicing) y las regiones codificantes del gen LDLR; la fase 2 consistió en el estudio de grandes rearreglos por la técnica de amplificación de sondas dependiente de ligandos múltiples (MLPA).
Presentación de los datosLos datos de los pacientes se muestran en tablas y gráficos y las características clínicas y de laboratorio se describen en forma individual.
ResultadosEn este trabajo se estudiaron 3.140 sujetos que asistieron a la unidad de endocrinología, de los cuales 10 (0,32%) tuvieron características clínicas y bioquímicas compatibles con HF. De estos 10 pacientes, 5 tuvieron diagnóstico clínico de HF probable y 5 de certeza (puntuación>8).
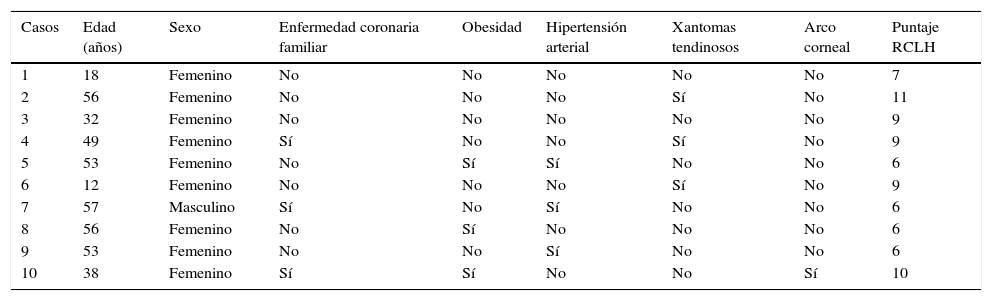
En la tabla 2 se presentan los datos clínicos de los 10 pacientes, cuyas edades estuvieron comprendidas entre los 12 y los 57 años. Todos, excepto uno, eran de sexo femenino. Tres pacientes tuvieron antecedente de algún familiar de primer grado con enfermedad coronaria prematura y ninguno comunicó antecedente personal de esta patología, ni de enfermedad arterial periférica o cerebrovascular. Con respecto a las comorbilidades, 3 pacientes tuvieron obesidad, 3 hipertensión arterial primaria y ninguno fue diabético. En relación con signos clínicos asociados a HF, 3 pacientes presentaban xantomas tendinosos y solo uno arco corneal. En la figura 1 se muestran los xantomas tendinosos del tendón de Aquiles en la paciente 2, de 56 años de edad, y el arco corneal en la paciente 10, de 38 años de edad.
Datos clínicos de los pacientes con hipercolesterolemia familiar
| Casos | Edad (años) | Sexo | Enfermedad coronaria familiar | Obesidad | Hipertensión arterial | Xantomas tendinosos | Arco corneal | Puntaje RCLH |
|---|---|---|---|---|---|---|---|---|
| 1 | 18 | Femenino | No | No | No | No | No | 7 |
| 2 | 56 | Femenino | No | No | No | Sí | No | 11 |
| 3 | 32 | Femenino | No | No | No | No | No | 9 |
| 4 | 49 | Femenino | Sí | No | No | Sí | No | 9 |
| 5 | 53 | Femenino | No | Sí | Sí | No | No | 6 |
| 6 | 12 | Femenino | No | No | No | Sí | No | 9 |
| 7 | 57 | Masculino | Sí | No | Sí | No | No | 6 |
| 8 | 56 | Femenino | No | Sí | No | No | No | 6 |
| 9 | 53 | Femenino | No | No | Sí | No | No | 6 |
| 10 | 38 | Femenino | Sí | Sí | No | No | Sí | 10 |
RCLH: Red de Clínicas de Lípidos de Holanda.
 Xantomas en el tendón de Aquiles en paciente femenino de 56 años de edad. B) Arco corneal en paciente femenino de 38 años de edad.")
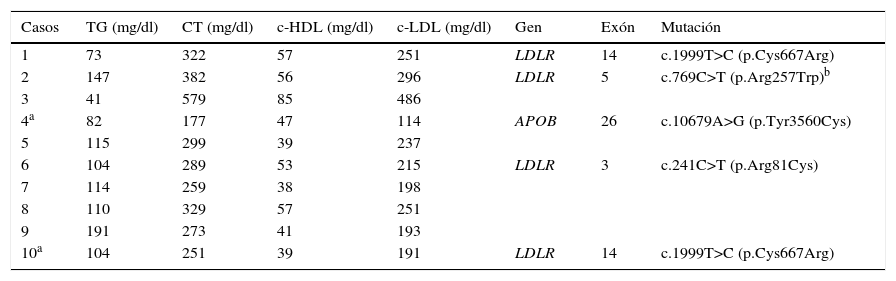
En la tabla 3 se muestran los valores de los lípidos y las mutaciones encontradas en los genes LDLR y APOB en los 10 pacientes con HF. A pesar de que 2 pacientes estaban recibiendo tratamiento con estatinas, solo uno de ellos (paciente núm. 4) presentó niveles aceptables de c-LDL (114mg/dl). El resto de los pacientes tuvieron valores claramente elevados de c-LDL, los cuales oscilaron entre 191 y 486mg/dl. La mayoría de los pacientes mostraron valores normales de triglicéridos y de c-HDL. En total se detectaron 4 mutaciones diferentes, 3 en el gen LDLR y una en APOB. La variante p.Cys667Arg fue la más común ya que se observó en 2 pacientes no relacionadas y las restantes 3 mutaciones p.Arg81Cys, p.Arg257Trp y p.Tyr3560Cys —esta última del gen APOB— solo se detectaron en un individuo. Considerando estos datos y asumiendo que la mutación p.Arg257Trp ha sido clasificada como no patogénica en un estudio funcional, en el 40% de los pacientes con HF se confirmó el diagnóstico molecular.
Perfil de lípidos y mutaciones en los genes LDLR y APOB en pacientes con hipercolesterolemia familiar
| Casos | TG (mg/dl) | CT (mg/dl) | c-HDL (mg/dl) | c-LDL (mg/dl) | Gen | Exón | Mutación |
|---|---|---|---|---|---|---|---|
| 1 | 73 | 322 | 57 | 251 | LDLR | 14 | c.1999T>C (p.Cys667Arg) |
| 2 | 147 | 382 | 56 | 296 | LDLR | 5 | c.769C>T (p.Arg257Trp)b |
| 3 | 41 | 579 | 85 | 486 | |||
| 4a | 82 | 177 | 47 | 114 | APOB | 26 | c.10679A>G (p.Tyr3560Cys) |
| 5 | 115 | 299 | 39 | 237 | |||
| 6 | 104 | 289 | 53 | 215 | LDLR | 3 | c.241C>T (p.Arg81Cys) |
| 7 | 114 | 259 | 38 | 198 | |||
| 8 | 110 | 329 | 57 | 251 | |||
| 9 | 191 | 273 | 41 | 193 | |||
| 10a | 104 | 251 | 39 | 191 | LDLR | 14 | c.1999T>C (p.Cys667Arg) |
APOB: gen de la apolipoproteína B; c-HDL: colesterol transportado por lipoproteínas de alta densidad; c-LDL: colesterol transportado por lipoproteínas de baja densidad; CT: colesterol total; LDLR: gen del receptor de lipoproteína de baja densidad; TG: triglicéridos.
Hasta donde tenemos conocimiento, este es el primer estudio en una unidad de endocrinología de Venezuela, en que, además de analizar la frecuencia de HF, se analizan las características clínicas, bioquímicas y moleculares de los sujetos afectos. Los resultados de este estudio sugieren que aproximadamente una de cada 314 personas que acuden a consulta de endocrinología presentan HF. La prevalencia de HF heterocigota en población general se ha estimado comúnmente en una de cada 500 personas14; sin embargo, reportes más recientes sugieren una frecuencia aún mayor de aproximadamente una de cada 200-300 personas3,4,8, similar a lo observado en este estudio.
Los xantomas tendinosos son patognomónicos de HF y se observaron en el 30% de los pacientes que analizamos. Se ha demostrado que los sujetos con xantomas tienen 3 veces mayor riesgo de enfermedad arterial coronaria en comparación con los que no los presentan15. A pesar de su valor clínico, se observan en menos del 30% de los casos, por lo que su ausencia no excluye el diagnóstico clínico16.
A nivel mundial la mayoría de los pacientes con HF están sin diagnosticar y por lo tanto sin tratamiento, o bien con tratamiento insuficiente8,17. La detección de HF cumple con los criterios de la Organización Mundial de la Salud (OMS) para el tamizaje sistemático de una enfermedad y es coste-efectiva para detectar nuevos casos de HF mediante el tamiz en cascada familiar a partir de la identificación de los casos índice12. En este estudio se evidenció que ninguno de los pacientes con HF había sido diagnosticado previamente y solo 2 de ellos recibían tratamiento hipolipemiante con estatinas pero sin alcanzar las metas de control terapéutico (c-LDL ¿100mg/dl). Se destaca que ninguno de los sujetos tratados recibía tratamiento combinado ni estatinas a dosis máxima, similar a lo descrito en un estudio observacional español con participación de médicos de atención primaria y atención especializada, en el cual se observó que menos del 5% de los casos con diagnóstico genético consiguen el objetivo en los niveles de c-LDL y menos del 15% de estos pacientes están recibiendo tratamiento combinado a dosis máxima16.
Los criterios de la RCLH fueron desarrollados con la finalidad de facilitar la selección de pacientes para el análisis genético como parte del programa nacional para el tamizaje de HF en Holanda11. Utilizando dichos criterios se observa que en el 40% de los pacientes seleccionados (diagnóstico de probabilidad+certeza) se logró establecer el diagnóstico clínico, bioquímico y molecular ya que se detectó la mutación causante de HF, siendo mayor la tasa de detección a medida que se incrementa la puntuación obtenida. Estos resultados son similares a los reportados en Dinamarca, donde se detectaron mutaciones en el 48,1% de los sujetos si se incluían aquellos con diagnóstico de probabilidad y de certeza, mientras que la tasa de detección aumentó al 62,9% si solo se consideraban los pacientes con diagnóstico de certeza18.
En concordancia con otros países de Latinoamérica y el mundo2,9, la mayor parte de las mutaciones encontradas en los pacientes con HF afectan el gen LDLR que se localiza en el brazo corto del cromosoma 19 y está compuesto por 18 exones19. Hasta la fecha se han descrito más de 1.800 variantes diferentes, no todas patogénicas20. De acuerdo con la ubicación de las mutaciones se han descrito 5 clases que afectan en diferentes sitios en la vía del receptor de c-LDL: clase 1 (ausencia de síntesis del receptor de c-LDL), clase 2 (defecto en el transporte del receptor), clase 3 (falla en la unión del c-LDL con su receptor), clase 4 (alteración en la internalización del complejo c-LDL/receptor de c-LDL) y clase 5 (disfunción en el reciclaje del receptor)19.
Un estudio previo realizado en Maracaibo, Venezuela, evaluó un total de 65 pacientes con niveles elevados de c-LDL y analizó el exón 4 del gen LDLR y las mutaciones R3500Q, R3500W y R3500C del gen APOB; ninguno de los sujetos estudiados presentaba xantomas tendinosos ni arco corneal y solo uno presentó xantelasma. En 5 pacientes fue posible encontrar una mutación en LDLR, 4 de ellas noveles y una descrita previamente en población alemana asociada también a hipercolesterolemia10. Se destaca que ninguna de estas mutaciones fue observada en pacientes de Ciudad Bolívar.
En los casos índice 1 y 10 se identificó la misma mutación (pacientes no relacionadas), un cambio de timina a citosina en el nucleótido 1999, cuyo resultado es la sustitución de cisteína por arginina en el codón 667 del exón 14 de LDLR. Esta mutación ha sido previamente descrita en miembros consanguíneos de una familia de Siria, donde se demostró que dicho cambio puede afectar el transporte del receptor de c-LDL entre el retículo endoplásmico y el aparato de Golgi21.
En el análisis molecular de la paciente 2 se identificó la variante c.769C>T, localizada en el exón 5 del gen LDLR. Dicha variante produce el cambio de arginina por triptófano en la posición 257 del receptor de c-LDL. Esta alteración resulta controversial, ya que ha sido descrita al menos en 3 ocasiones en familias con HF22-24; sin embargo, estudios funcionales recientes reportaron que se trata de una variante no patogénica25. Por su parte, en la paciente 6 se identificó la mutación c.241C>T, localizada en el exón 3 del gen LDLR, que genera el cambio de arginina por cisteína en la posición 81 del receptor de c-LDL. Esta mutación en sentido equivocado ha sido descrita previamente y afecta el dominio de unión a ligando del receptor26. Esta fue la única paciente incluida en el estudio con menos de 18 años de edad y se sospechó el diagnóstico de HF debido a la presencia de c-LDL >190mg/dl, como sugieren algunas guías de diagnóstico y tratamiento11,12.
En el 5% de los pacientes con diagnóstico de HF se encuentra una mutación en el gen APOB27. Esta condición también es conocida como apolipoproteína B defectuosa familiar y produce un fenotipo indistinguible de HF. En la paciente 4 se detectó la mutación c.10679A>G, localizada en el exón 26 del gen APOB, que produce el cambio de tirosina por cisteína en la posición 3560 de la apolipoproteína B. Esta mutación también ha sido descrita previamente28 y afecta la unión de las partículas de c-LDL con su receptor, lo que genera aumento en las concentraciones plasmáticas de c-LDL.
En los pacientes con diagnóstico clínico de HF en los que no se detecta ninguna mutación el aumento en la concentración plasmática de c-LDL puede deberse a causas poligénicas, es decir, a un acúmulo de variantes genéticas que aumentan el c-LDL y pueden semejar un fenotipo de HF29. Otra posible explicación es la incapacidad de las técnicas actuales para detectar todas las mutaciones, por ejemplo, mutaciones intrónicas en el LDLR, mutaciones en PCSK9 o mutaciones en otros genes no descubiertos causantes de HF30. Se destaca que en este estudio no se analizó el gen PCSK9, pero, independientemente del hallazgo de mutaciones, el diagnóstico de HF no debe ser descartado ya que en un número significativo de casos en los que se han estudiado los genes principales que causan HF no se logran identificar defectos monogénicos30. Además, todos los pacientes deben ser adecuadamente tratados debido a que la mayor parte de los estudios de morbimortalidad se han realizado en pacientes con diagnóstico clínico y no con diagnóstico genético31,32. A pesar de ello, se ha demostrado que para cualquier nivel de colesterol c-LDL los portadores de mutación genética detectada tienen un riesgo aumentado de enfermedad arterial coronaria33.
Los pacientes con HF tienen un riesgo elevado de presentar enfermedades cardiovasculares, sin embargo, dicho riesgo puede modificarse con la presencia o no de otros factores concomitantes como la diabetes mellitus, la hipertensión arterial y la obesidad8,12. Podemos destacar que ninguno de los pacientes analizados con HF presentaba antecedentes personales de enfermedad arterial coronaria ni tampoco de enfermedad cerebrovascular; esto pudiera deberse a la presencia de factores favorables como sexo femenino (90%), edad menor a 40 años (40%) y ausencia de tabaco en todos los participantes.
Limitaciones del estudioAunque este estudio provee hallazgos de interés, es necesario reconocer algunas limitaciones: 1) la frecuencia de HF ha sido determinada en sujetos que acuden a una unidad de endocrinología, por lo que estos resultados pudieran no representar a la población general de Ciudad Bolívar ni a la de Venezuela; sin embargo, muchos estudios de prevalencia disponibles en la literatura están basados en registros hospitalarios, pacientes hospitalizados e incluso cálculos empleando la ecuación de Hardy-Weinberg y la frecuencia estimada de HF homocigota2,30; 2) algunos pacientes que acuden a consulta reciben tratamiento hipolipemiante y en ausencia de historia familiar o signos clínicos de hipercolesterolemia el diagnóstico de HF resulta más complejo. Por tanto, es posible que con este método de tamizaje algunos sujetos con HF no hayan sido seleccionados.
ConclusionesEn conclusión, la HF es un problema de salud pública mundial, tanto por su frecuencia como por la gravedad de sus consecuencias. En esta Unidad de Endocrinología de Ciudad Bolívar-Venezuela se encontró una frecuencia de HF de uno por cada 314 pacientes, lo cual sugiere que existe un subregistro importante de esta enfermedad y hace evidente la deficiencia que existe en nuestro sistema de salud en la detección de pacientes con HF. Las mutaciones que afectan al gen LDLR son las principales causantes de HF en este grupo de pacientes.
Conflicto de interesesLos autores declaran que al momento de escribir el manuscrito no existen conflictos de intereses.
