






Se presenta una revisión de los métodos que se han desarrollado dentro de la Química Teórica para estudiar a la materia en diversas escalas de espacio y tiempo. La trayectoria de un conocido químico teórico se utiliza para mostrar cómo, en 4 décadas, los desarrollos de teorías, programas (algoritmos) y de la tecnología computacional nos permiten en la actualidad el estudio confiable de la estructura electrónica de sistemas cuyos tamaños los hacían prohibitivos hace 40 años. Se señala y enfatiza que la dificultad central en el estudio teórico de la Química reside en que, en todas los escalas, se trata de un problema de muchos cuerpos cuya solución y comprensión siguen siendo un reto científico no resuelto y fascinante.
A review of the methods developed within Theoretical Chemistry to study matter in many spatial and temporal scales is presented. The scientific career of a well known theoretical chemist is used to show how the astonishing advances that have been achieved in four decades in the development of theories, codes (algorithms) and computer technology allow nowadays the reliable study of the electronic structure of systems whose sizes were unmanageable forty years ago. It is emphasized that the central difficulty in studying Chemistry from a theoretical perspective is that, in every scale, one deals with a many-body problem whose solution and comprehension are still an unsolved and fascinating scientific challenge.
Uno de los paradigmas centrales de la Química es la relación que existe entre la estructura molecular (la disposición espacial de los átomos en las moléculas) y la función de las moléculas. Por esta razón, los químicos dedican grandes esfuerzos en tratar de conocer la estructura de las moléculas para, con esta valiosa información, intentar racionalizar el comportamiento químico y físico de los compuestos. La razón última por la cual los átomos de una molécula adoptan una cierta disposición en el espacio se debe a las interacciones existentes a nivel microscópico. Estas interacciones tienen origen en la estructura electrónica de las moléculas a la cual tenemos acceso tanto experimental como teóricamente. La herramienta experimental para escudriñar la estructura electrónica de la materia es la espectroscopia, particularmente la ultravioleta-visible (UV-Vis), la de rayosX y, más recientemente, las elegantes y fascinantes espectroscopias de femto- y ato-segundos, así como las de una molécula (single molecule spectroscopy). Desde el punto de vista teórico, la herramienta para estudiar la estructura electrónica molecular es la Mecánica Cuántica, que es la rama de la Física que nos permite explorar el comportamiento del mundo microscópico en escalas inaccesibles a los sentidos humanos, entre ellas, la escala electrónica. La Mecánica Cuántica quedó bien establecida entre los años 1925 y 1926, después de que se demostró que sus distintas formulaciones (la matricial de Heisenberg, la ondulatoria de Schrödinger y la algebraica de Dirac) están relacionadas. Casi inmediatamente después de que se establecieron los postulados de la Mecánica Cuántica, se inició un alud de aplicaciones a una pléyade de problemas de interés que ansiosamente esperaban ser estudiados, entre ellos, el de la estructura electrónica de la materia. Dado que el comportamiento de la materia en las condiciones donde existen los átomos y las moléculas (recuerde el lector que a las condiciones de temperatura y presión prevalecientes al inicio del Universo o en la mayoría de las estrellas, la energía es tan elevada, que los átomos y las moléculas se desintegran en partículas elementales como neutrones, protones y electrones), se rige por las interacciones eléctricas y magnéticas, entonces el camino formal y riguroso que deberíamos tomar para estudiar la estructura electrónica molecular debería ser la Electrodinámica Cuántica. Destacamos la importancia de la Electrodinámica Cuántica señalando que el espín electrónico, propiedad física imprescindible para entender el ordenamiento de los átomos en la tabla periódica, la naturaleza del enlace químico o el comportamiento magnético de la materia, tiene su origen riguroso y formal en la descripción relativista de la Mecánica Cuántica. Hoy sabemos que la Mecánica Cuántica no-relativista aplicada a un conjunto de electrones y núcleos es un excelente punto de partida para iniciar nuestra exploración del mundo de la estructura electrónica molecular, donde el reto no es descubrir nuevas interacciones o nuevas partículas, sino el hecho de que el problema, aun el más simple, es un problema de muchos cuerpos. Este es el gran reto de la Química Teórica y, probablemente, lo que motivó a Dirac su famosa frase de 1929, que tanto ha irritado —e irrita— a muchos químicos, y que dice: «Así, las leyes físicas subyacentes necesarias para tener una teoría matemática para gran parte de la Física y toda la Química son completamente conocidas,…» (Dirac, 1929). Sin la menor duda, la afirmación es contundente pero, en descarga de Dirac, hay que señalar que no siempre se continúa la lectura del párrafo que dice: «… y la dificultad reside solamente en que la aplicación exacta de estas leyes conduce a ecuaciones demasiado complicadas para ser solubles. Por tanto, es deseable desarrollar métodos aproximados prácticos para aplicar la Mecánica Cuántica, lo cual llevará a una explicación de las características principales de sistemas atómicos complejos sin demasiado cómputo» (Dirac, 1929). La claridad en la visión de Dirac respecto al reto es sorprendente y sus afirmaciones nos sirven para empezar a entender la razón por la cual nuestra actual comprensión de la estructura electrónica de la materia está íntimamente ligada al desarrollo tanto de la tecnología computacional en «fierros» (hardware), como en algoritmos y métodos (software), con los cuales podemos intentar entender la Química desde un punto de vista microscópico, es decir, electrónico.
La postura de Dirac, un poco pesimista, respecto a la imposibilidad de resolver las ecuaciones «exactas» de la Mecánica Cuántica aplicadas al problema de la estructura electrónica de la materia está ampliamente rebasada. De hecho, en las décadas de los 50 y 60 del siglo pasado se empezaron a desarrollar métodos y programas para calcular la estructura electrónica que se denominaron, con toda intención, métodos ab initio, que en latín literalmente significa desde el principio o que, como lo entiende la comunidad de la Química Teórica y Computacional, a primeros principios. Esta denominación la acuñó el profesor Robert G. Parr en un heroico trabajo sobre la predicción de los primeros estados excitados de la molécula de benceno (Parr, Craig y Ross, 1950). La motivación principal de utilizar este nombre es precisamente enfatizar que la única información que se le proporciona a estos métodos para resolver las ecuaciones del problema electrónico son el número de electrones, el número de núcleos, la carga nuclear de cada núcleo y su correspondiente posición en el espacio. Para dejar una idea más clara y concreta de los métodos existentes para estudiar la estructura electrónica de la materia, en la tabla 1 se presenta un cuadro esquemático con las metodologías teóricas, las cuales explicaremos brevemente a continuación (Szabo y Ostlund, 1996; Schatz y Ratner, 2002; McQuarrie, 2007; Atkins y Friedman, 2011; Levine, 2013).
Métodos teóricos para estudiar la estructura electrónica de la materia
| Tipo | Descripción | Ejemplos |
|---|---|---|
| A primeros principios ab initio | Lo único que tenemos que dar como información son el número de átomos, de electrones, el tipo de átomos y las posiciones de los núcleos en el espacio | Vía la función de onda Ψx1,x2,…,xN |
| Vía la densidad electrónica ρ(r) | ||
| Semiempíricos | Realizan aproximaciones a las ecuaciones ab initio, lo que introduce parámetros los cuales se fijan minimizando errores respecto a algunas propiedades experimentales. Se tienen que parametrizar todos los átomos y pares de átomos | PPP, CNDO, CINDO, MINDO, AM3, SINDO, ZINDO, MOPAC … |
| Hamiltonianos modelo | Proponen hamiltonianos con formas específicas que intentan describir problemas electrónicos particulares | Hückel extendido (banda rígida), hamiltoniano de Hubbard, teoría BCS… |
Como ya se dijo en el párrafo anterior, jerárquicamente tenemos a la cabeza los caminos que recurren al menor número de aproximaciones. Estos son los denominados métodos a primeros principios o ab initio. La Mecánica Cuántica establece que el objeto portador de «toda» la información dinámica de un sistema es la función de onda, Ψx1,x2,,xi,xN;R1,R2,,RARM, que es una función compleja, en el sentido matemático, y complicada, en el sentido estricto, y que depende de las coordenadas de los N electrones y los M núcleos presentes en el sistema. Las coordenadas electrónicas, xi, i=1, …, N, contienen tanto información espacial como sobre el espín del electrón correspondiente, mientras que las nucleares, RA, A=1, …, M, solamente describen la posición de cada núcleo en el espacio tridimensional. La Mecánica Cuántica también nos señala que para conocer la función de onda de un sistema tenemos que resolver la ecuación de Schrödinger dependiente del tiempo, que a su vez depende del hamiltoniano del problema (Szabo y Ostlund, 1996; Schatz y Ratner, 2002; McQuarrie, 2007; Atkins y Friedman, 2011; Levine, 2013). Si nos concentramos en problemas estáticos y no-relativistas, el problema deja de tener la dificultad de describir la evolución temporal de la función de onda; en este caso tenemos que resolver la conocida como ecuación de Schrödinger independiente del tiempo, que tampoco resulta ser muy fácil de resolver. Para resolverla tenemos que recurrir a la aproximación de Born-Oppenheimer que al considerar que los núcleos tienen masas mucho mayores (aproximadamente, y cuando menos, 1,000 veces más grandes) que los electrones, nos permite suponer que los núcleos están fijos, lo que lleva a la posibilidad de separar el movimiento electrónico del nuclear. La ecuación resultante es una ecuación de Schrödinger que solo se «fija» en las coordenadas electrónicas y lo hace a través de la que denominamos función de onda electrónica que depende «explícitamente» de las coordenadas electrónicas y «paramétricamente» de las posiciones espaciales de los núcleos. La dependencia paramétrica implica que para obtener esta función de onda tenemos que proporcionar las posiciones y cargas de los núcleos, pero no tenemos que preocuparnos por describir el movimiento nuclear. Sin duda, el problema a resolver es menos complejo que el original: en el problema original tenemos que obtener una función que depende de 4N+3M variables (recuerde que N es el número de electrones y M el de núcleos), mientras que con la aproximación de Born-Oppenheimer tenemos que encontrar una función que «únicamente» depende de 4N variables, las coordenadas electrónicas.
Pero la Naturaleza nos reserva una complicación adicional. En los años 40 se demostró que como consecuencia de una simetría fundamental en los sistemas cuánticos de muchos cuerpos, en el intercambio de las coordenadas de cualquier par de partículas presentes en el sistema, la función de onda tiene que pertenecer a uno de los siguientes tipos: o cambia de signo o no cambia de signo. A la primera la llamamos función de onda simétrica y a la segunda antisimétrica. Esto tiene consecuencias trascendentales pues implica que en la Naturaleza existen 2 grandes tipos de sistemas de muchas partículas: los que se rigen por funciones de onda simétricas y a cuyas partículas denominamos «bosones», y los que lo hacen por funciones de onda antisimétricas y que llamamos «fermiones». Los electrones pertenecen a esta última categoría y, por consiguiente, su dinámica cuántica se rige por funciones de onda antisimétricas. Antes de retomar el punto sobre la estructura electrónica es importante señalar que el comportamiento de la función ante el intercambio también señala una diferencia en los espines y estadísticas que siguen los sistemas, teorema de la Mecánica Cuántica que se conoce como teorema de espín-estadística y fue demostrado por Markus Fierz y Wolfang Pauli entre 1939 y 1940. Los bosones son partículas con espines enteros 1,2,3, y siguen la estadística de Bose-Einstein, y los fermiones tienen espines semienteros 12,32,52, y siguen la estadística de Fermi-Dirac. Una consecuencia de la antisimetría que no podemos dejar de mencionar es que es la responsable del famoso principio de exclusión de Pauli, el cual establece que la probabilidad de encontrar a 2 electrones con el mismo espín y en el mismo punto del espacio es nula. Es ilustrativo demostrar este principio para el caso de un sistema con 2 electrones. En este caso, la función de onda electrónica está dada por ψr1σ1,r2σ2 donde ri. es el vector de posición del i-ésimo electrón y σi es su correspondiente coordenada de espín, la cual solamente puede tomar 2 valores que se suelen denotar como espín arriba (↑) o espín abajo (↓). Si tenemos 2 electrones con el mismo espín, σ1=σ2=σ, la condición de antisimetría establece que ψr1σ,r2σ=−ψr1σ,r2σ, la cual si rearreglamos se puede escribir como ψr1σ,r2σ+ψr1σ,r2σ=0. Esta igualdad no tiene mayor problema si las posiciones espaciales de los 2 electrones son distintas, esto es, si r1≠r2, pero, si las 2 posiciones espaciales son iguales, r1=r2=r, entonces la ecuación se reduce a 2ψrσ,rσ=0. Ahora, como el cuadrado de la función de onda nos da la probabilidad de encontrar al sistema en esa situación en particular, la última ecuación claramente establece que la probabilidad de encontrar a 2 electrones con el mismo espín y en la misma posición en el espacio es nula. Esta es la mejor manera de entender el origen del principio de exclusión de Pauli y pido al lector que aprecie que en esta argumentación no se tuvo que recurrir a la manera más común como se nos enseña este fundamental principio de la Mecánica Cuántica en términos de llenado de orbitales atómicos.
Si retomamos nuestra discusión de cómo calculamos la estructura electrónica de la materia, dentro de la aproximación de núcleos fijos (Born-Oppenheimer), no relativista y en estados estacionarios, lo que tenemos que resolver es la ecuación de Schrödinger independiente del tiempo pero, por lo expuesto en el párrafo anterior, lo tenemos que hacer imponiendo la condición de que la función de onda del sistema sea antisimétrica ante la permutación (intercambio) de cualquier par de electrones. Una manera de incorporar esta trascendental condición es por medio de determinantes que en la teoría cuántica de estructura electrónica conocemos como determinantes de Slater. Puede el lector entender la razón de usar estos objetos matemáticos al recordar que una propiedad básica de los mismos es que cuando se intercambia cualquier par de filas o columnas, el valor del determinante cambia de signo. Un determinante de Slater corresponde a una configuración electrónica específica del sistema. El usar un determinante de Slater da origen al nivel ab initio más básico, que conocemos como la teoría de Hartree-Fock (HF), en honor de los físicos Douglas R. Hartree y Vladimir Fock, quienes desarrollaron esta teoría allá en los años 30 del siglo pasado. La teoría de HF es el primer peldaño ab initio para la descripción cuántica de la estructura electrónica por medio de la función de onda. La capacidad predictiva de HF es aceptable pero no suficiente para poder hacer inferencias químicas confiables. ¿Qué le falta a HF? En la teoría de estructura electrónica, no relativista y dentro de la aproximación de Born-Oppenheimer, a todo lo que le falta a HF para proporcionar el resultado exacto lo llamamos «correlación electrónica». Este término implica que hay interacciones entre los electrones que no se toman en cuenta en la teoría de HF. Al tomarlas en cuenta decimos que estamos incluyendo la correlación electrónica. Así, la descripción completa y exacta de un átomo, molécula o sólido requiere de ir más allá de la aproximación en la cual la función de onda se describe por solamente un determinante de Slater. Esto se aprecia claramente si consideramos la manera en que podemos escribir exactamente la función de onda de un sistema:
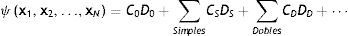
En esta ecuación, estamos considerando la función de onda estacionaria de un sistema con N electrones, cada uno de ellos descrito por el tetra-vector xi=riσi. D0 representa al determinante de Slater con el que se hace la primera aproximación que corresponde a HF; las sumas que aparecen en el lado derecho de la ecuación (1) incluyen los determinantes que corresponden a lo que conocemos como las excitaciones simples, dobles, etc. Esta es una manera de incorporar la correlación electrónica. Los coeficientes C0, CS, CD, se determinan recurriendo a uno de los métodos aproximados que tiene la Mecánica Cuántica para resolver la ecuación de Schrödinger; nos referimos al método variacional, el cual busca los valores de los coeficientes que minimizan la energía total del sistema. Como cada determinante D0, DS, DD, corresponde a una configuración electrónica diferente, a los métodos que determinan la estructura electrónica de esta manera se les conoce como métodos de interacción de configuraciones (CI, por sus siglas en inglés de configuration interaction). El punto donde se corta o termina la suma de la ecuación (1) determina el tipo de método. Así, si nos quedamos solamente con las configuraciones que se obtienen de «mover» un electrón, las llamadas excitaciones simples, el método se llama CI-S, o si incluimos excitaciones simples y dobles, entonces se denomina CI-SD. Como podrá anticipar el lector, conforme se incluyen más términos en la suma, el cálculo se vuelve más demandante. Una manera de estimar cuán demandante es un método de estructura electrónica es por medio de lo que se denomina «escalamiento formal», que es la potencia a la que se tiene que elevar el número de electrones N y corresponde al esfuerzo computacional que se tiene que realizar en el paso más demandante en el proceso de resolución de la ecuación de Schrödinger. Así, el método de HF tiene un escalamiento formal de N4, mientras que CI-SD tiene un escalamiento formal de N6. Basta un ejemplo para apreciar el significado práctico (computacional) del escalamiento formal. Si tenemos una molécula como el agua que tiene 10 electrones, un cálculo de CI-SD requiere un esfuerzo computacional 100 veces mayor que HF o para una molécula como el fulereno, C60, que tiene 360 electrones, CI-SD sería ¡129,600 veces más costoso que HF! En suma, la correlación, por este camino, es extremadamente cara.
Como señalamos anteriormente, y se muestra en la tabla 1, existe otra manera ab initio de incluir la correlación electrónica y es por medio de la teoría de funcionales de la densidad (DFT, por sus siglas en inglés de density functional theory) (Parr y Yang, 1994; Fiolhais, Nogueira y Marques, 2010; Engel y Dreizler, 2011). Históricamente, los orígenes de la DFT se remontan a los trabajos de Fermi y de Thomas quienes entre 1928 y 1929 mostraron que haciendo uso de la estadística que siguen los electrones (fermiones), la estadística de Fermi-Dirac que discutimos arriba, se puede desarrollar un modelo del átomo capaz de predecir, con sorprendente cercanía, los valores experimentales de las energías totales de los átomos en la tabla periódica. A este modelo se le conoce como el modelo atómico de Thomas-Fermi y se considera el primer funcional de la densidad en la historia de la estructura electrónica de la materia. Por casi 4 décadas, los métodos que siguieron el espíritu del modelo de Thomas-Fermi, a los que se les denominó modelos estadísticos, se limitaron a ser una curiosidad fisicomatemática y fueron explotados en ramas de la Física, como la Astrofísica, donde se usaron para calcular diversas propiedades de estrellas de neutrones, por ejemplo. La Química descartó estos métodos por 2 grandes razones: primero, los modelos estadísticos son incapaces de predecir la estructura de capas de los átomos y, en segundo lugar, son incapaces de predecir la existencia de una molécula, pues todos estos métodos predicen que la energía de una especie molecular formada por los átomos A y B, la especie AB, tiene un valor «mayor» que la de los átomos constituyentes, esto es,
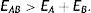
Por supuesto, estos resultados desmotivaron, durante décadas, la exploración de la estructura electrónica por medio de los métodos estadísticos. La situación cambió drásticamente en 1964 y 1965. Primero, en 1964 Walter Kohn y Pierre Hohenberg demostraron 2 teoremas que hoy son las piedras angulares de la DFT (Parr y Yang, 1994; Fiolhais et al., 2010; Engel y Dreizler, 2011). El primer teorema establece que para conocer la energía del estado basal (el estado basal es el de menor energía de un sistema) de un sistema de fermiones no es necesario calcular la función de onda, Ψx1,x2,xN, sino que basta con obtener su densidad electrónica, ρ(r). Este teorema tiene enormes implicaciones conceptuales y prácticas, tanto en la estructura electrónica de la materia como, en general, en la Mecánica Cuántica de muchos cuerpos. Para apreciar esto en toda su magnitud considere el lector que la función de onda electrónica es un objeto matemático complejo y complicado «que depende de 4N variables», 3 espaciales y una de espín por cada electrón, mientras que la densidad electrónica es un campo escalar «que solamente depende de 3 variables», las variables espaciales. Como parte de este primer teorema también se demuestra que la energía del estado basal del sistema, Eρ, se puede escribir como
donde vr es el potencial externo, que en un átomo es el potencial debido al núcleo y en una molécula el debido a todos los núcleos presentes en la misma; el otro ingrediente que aparece en la ecuación (3), Fρ, es de importancia central en la DFT. Se le conoce como el «funcional universal» y contiene la contribución de energía cinética y «toda» la contribución a la energía debida a la interacción entre los electrones, la cual, claro está, contiene toda la correlación de la que hablamos arriba. A este funcional se le llama universal pues de lo único que depende es de la densidad electrónica del sistema y no tiene ninguna dependencia «explícita» en la naturaleza específica del mismo; en otras palabras, no depende, explícitamente, del potencial externo. Dado un potencial externo (tipo de sistema) y el número de electrones, el segundo teorema de Hohenberg y Kohn nos proporciona el camino para encontrar la densidad del estado basal: se debe minimizar la energía de la ecuación (3) sujeta a la restricción de que la densidad integre al número correcto de electrones, es decir, el segundo teorema es un principio variacional o un proceso de optimización. Lo dicho hasta ahora parece ser maravilloso, pero tenemos un gran «pero». A diferencia del camino de la función de onda del que conocemos el ingrediente principal de la ecuación de Schrödinger, el hamiltoniano, en la DFT, hasta la fecha, no se conoce la forma exacta del funcional universal. La situación es tan grave que de no ser por el siguiente gran paso que dieron Kohn y Sham (KS) en el año 1965, la DFT se habría convertido en una más de las teorías de la Física que no pueden usarse en aplicaciones concretas como las que nos interesan en la Química. No es el objetivo ni tenemos el espacio para explicar con detalle los fundamentos de la teoría de KS. Baste señalar que la genialidad de estos investigadores fue el introducir una referencia, un sistema de electrones no-interactuantes con la misma densidad que la del sistema completamente interactuante. La magia de esta consideración es que nos permite escribir de manera exacta la energía cinética y nos deja solamente un ingrediente desconocido, la llamada energía de intercambio-correlación. En muy poco tiempo, los físicos de materia condensada apreciaron la importancia de la teoría de KS y rápidamente se empezaron a desarrollar programas para calcular la estructura electrónica de sólidos y superficies. Los resultados en el campo de la interacción de átomos y moléculas con superficies fue particularmente exitosa, de manera que, para finales de los años 70, no quedaba la menor duda que el camino para estudiar este tipo de problemas era por medio de la DFT. El impacto de la DFT en la Química, particularmente de la teoría de KS, tuvo que esperar hasta finales de los 80 cuando Axel Becke utilizó su funcional de intercambio (Becke, 1993) junto con el funcional de correlación propuesto por Chentee Lee, Weitao Yang y Robert Parr (Lee, Yang y Parr, 1988), y demostró que con un costo computacional igual al requerido por HF, la teoría de KS proporcionaba resultados con calidades similares a las de métodos altamente correlacionados. El paso estaba dado y bastaron unos cuantos meses para que los grandes desarrolladores de códigos de estructura electrónica molecular, como John Pople y Nicholas Handy, implementaran el método de KS en sus programas. La revolución en la teoría de estructura electrónica había comenzado y nada la detendría. El éxito de sus predicciones y su consecuente impacto en la Química quedaron ampliamente reconocidas al otorgárseles, en 1998, el Premio Nobel de Química a Walter Kohn, por su desarrollo de la DFT, y a John Pople, por haber desarrollado los métodos que nos permiten calcular la estructura electrónica molecular tanto por el camino de la función de onda como por el de la densidad electrónica (Pople es el padre del conocido programa Gaussian).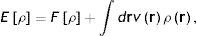
Por cualquiera de los caminos para estudiar de manera ab initio la estructura electrónica de la materia, las ecuaciones que tenemos que resolver son conjuntos de ecuaciones integrodiferenciales acopladas. En términos generales, estas ecuaciones se pueden resolver mediantes la introducción de una base o de una malla en el espacio tridimensional. En el primer caso, el problema se transforma en un problema matricial en el que los cuellos de botella computacionales son la construcción de la matriz (de Fock, por el camino de la función de onda, o de KS) y la diagonalización de la matriz de Fock o de KS. La etapa más demandante en la construcción de la matriz es la evaluación de las integrales de repulsión electrónica (ERIS, por sus siglas en inglés de electron repulsion integrals), las cuales tienen la siguiente forma general:
donde las funciones χμr son precisamente las funciones de base que suelen ser funciones centradas en los átomos (núcleos), r1 y r2 son los vectores de posición de 2 puntos en el espacio tridimensional y r12=r1−r2 es la distancia que los separa. Los índices μ,v,σ y λ son números cuánticos que distinguen a las funciones de base. De esta forma, de acuerdo con la ecuación (4), si un sistema se está calculando con NB funciones de base entonces se tienen que evaluar NB4 ERIS. Volvamos a los ejemplos que discutimos párrafos arriba. En lo que se llama «base mínima», que es la menor base con la cual se puede realizar un cálculo ab initio, la molécula de agua tiene 7 funciones de base (se incluyen los orbitales 1s, 2s, 2px, 2py, 2pz del oxígeno y los orbitales 1s de los hidrógenos) y el fulereno tiene 300 funciones de base (5 por cada átomo de carbono); de esta manera, en el agua hay que evaluar 2,401 integrales bielectrónicas y en C60 ¡8,100,000,000! Para apreciar lo que este número representa, si quisiéramos almacenar estas ERIS en disco, el archivo de integrales ocuparía, aproximadamente, medio Terabyte de espacio. En los últimos 40 años se han hecho grandes esfuerzos para manejar estos volúmenes de datos de manera óptima, explotando los avances en las arquitecturas de las computadoras y desarrollando algoritmos que evitan su almacenamiento.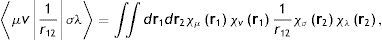
Para apreciar más claramente la demanda computacional de los métodos ab initio de estructura electrónica, en la tabla 2 se presentan los escalamientos formales de diferentes métodos. La última columna corresponde a una estimación del error promedio que se tiene con cada método en la predicción de la energía de enlace. Los valores no son totalmente precisos, pero muestran que para lograr mejores resultados por medio de la función de onda se requiere de enormes recursos computacionales.
Escalamientos formales de distintos métodos ab initio de estructura electrónica
| Métodos | Escalamiento | Errores promedio en kcal/mol |
|---|---|---|
| HF | N4 | >10.0 |
| DFT(KS) | N4 | 1.0-5.0 |
| CISD (excitaciones simples y dobles) | N6 | 1.7-5.8 |
| CISDT (excitaciones simples, dobles y triples) | N8 | 0.8 |
| CISDTQ (excitaciones simples, dobles, triples y cuádruples) | N10 | 0.01 |
El desarrollo de mejores métodos teóricos y de mejores algoritmos, así como el impresionante progreso en nuestra época de los recursos computacionales han impactado, sin el menor asomo de dudas, en la calidad de las predicciones, así como en el tamaño de los sistemas moleculares para los que se puede estudiar su estructura electrónica. Podemos apreciar este impacto siguiendo en el tiempo algunos de los trabajos de un químico teórico bien conocido. Usaré como referencia el trabajo de casi 5 décadas del profesor Henry F. Schaefer III. Como se puede consultar en Rothenberg, Young y Schaefer (1970), Schreiner, Severance, Jorgensen, Schleyer y Schaefer (1995) y Wang et al. (2013), en un periodo de 43 años la Química Teórica de frontera pasó de realizar estudios con geometrías fijas y usando el método ab initio más «simple» (HF), a estudiar sistemas en los que se optimiza la geometría de moléculas que contienen más de un centenar de átomos y 486 electrones. Podemos resumir diciendo que en los últimos 40 años, los desarrollos teóricos y computacionales nos permiten estudiar la estructura electrónica y molecular de manera ab initio en moléculas ¡30 veces! más grandes que las que se podían estudiar a principios de la década de los 70.
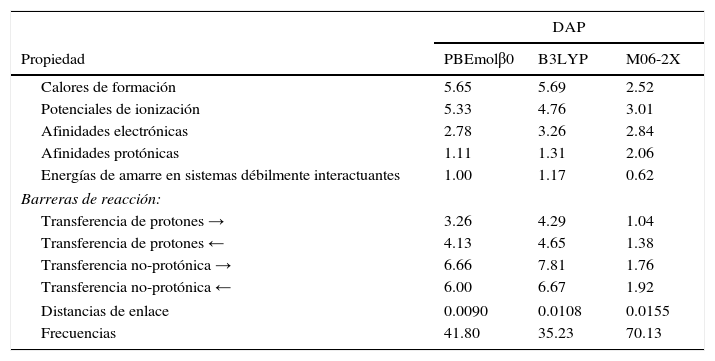
En la actualidad la teoría tiene una muy aceptable capacidad de predicción de propiedades relevantes para el químico, físico o para cualquier otro investigador o profesional quien necesite conocer alguna propiedad relacionada con la estructura electrónica de la materia. La confianza que se puede tener se ilustra en la tabla 3, donde se reportan 3 bloques de propiedades. El primero corresponde a una propiedad termodinámica muy importante, el calor o entalpía de formación, seguido de 2 diferencias de energía que involucran la transferencia de un electrón; el primero corresponde a la energía asociada con la extracción de un electrón (el potencial de ionización) y el segundo a la ganancia de un electrón (la afinidad electrónica); la cuarta cantidad, la afinidad protónica, corresponde a la diferencia de energía que existe cuando una especie química acepta un protón. La última de este bloque corresponde a sistemas que tienen energías de interacción muy pequeñas, como puentes de hidrógeno, interacciones π−π, de Van der Waals, etc. Las desviaciones absolutas promedio que se reportan son el promedio del valor absoluto de la diferencia entre el valor predicho por la teoría y el correspondiente valor experimental o de referencia. El segundo bloque, que se rotula como «barreras de reacción», corresponde a las diferencias de energía que existen entre los reactivos y el estado de transición (reacción a la derecha denotado por →), y los productos y el estado de transición (reacción a la izquierda denotado por ←). Estas barreras se reportan para reacciones en las que se lleva a cabo la transferencia de un protón o de otro tipo de átomo. Finalmente, en el último bloque se reportan propiedades estructurales como son las distancias de enlace y las frecuencias normales de vibración. Para cada propiedad se utilizan conjuntos de moléculas para las que se conocen con precisión los datos experimentales o de referencia.
Capacidades predictivas de algunos métodos ab initio de estructura electrónica
| DAP | |||
|---|---|---|---|
| Propiedad | PBEmolβ0 | B3LYP | M06-2X |
| Calores de formación | 5.65 | 5.69 | 2.52 |
| Potenciales de ionización | 5.33 | 4.76 | 3.01 |
| Afinidades electrónicas | 2.78 | 3.26 | 2.84 |
| Afinidades protónicas | 1.11 | 1.31 | 2.06 |
| Energías de amarre en sistemas débilmente interactuantes | 1.00 | 1.17 | 0.62 |
| Barreras de reacción: | |||
| Transferencia de protones → | 3.26 | 4.29 | 1.04 |
| Transferencia de protones ← | 4.13 | 4.65 | 1.38 |
| Transferencia no-protónica → | 6.66 | 7.81 | 1.76 |
| Transferencia no-protónica ← | 6.00 | 6.67 | 1.92 |
| Distancias de enlace | 0.0090 | 0.0108 | 0.0155 |
| Frecuencias | 41.80 | 35.23 | 70.13 |
Desviaciones absolutas promedio (DAP) de algunas propiedades calculadas con algunos funcionales híbridos. Todas las energías están en kcal/mol, las distancias en Å (1Å=1angstrom=10–10m) y las frecuencias en cm–1.
Fuente: datos tomados de Del Campo et al. (2012).
Los errores promedio que se reportan en la tabla 3 se calcularon por medio de la teoría de KS con 3 funcionales diferentes. El denominado PBEmolβ0 (Del Campo, Gazquez, Trickey y Vela, 2012) es un funcional desarrollado por nuestro grupo de investigación y M06-2X es un funcional desarrollado por el grupo del profesor Donald Truhlar de la Universidad de Minnesota. Es importante señalar que los parámetros existentes en el funcional propuesto por nosotros se fijaron recurriendo a condiciones físicas y/o matemáticas exactas, mientras que en los otros 2, sus parámetros se obtuvieron de minimizar el error con respecto a alguna propiedad específica. Por la manera de parametrizar, a funcionales como PBEmolβ0 se les conoce como funcionales «no-empíricos», mientras que a funcionales como B3LYP y M06-2X se les llama «empíricos». Como se puede observar en la tabla 3, el funcional que tiene la menor desviación respecto a los valores experimentales es el funcional M06-2X. Las únicas propiedades energéticas donde se ve superado por nuestro funcional es en las afinidades electrónicas y protónicas. En las propiedades estructurales también es ligeramente superado por nuestro funcional. Más que señalar la capacidad de nuestro funcional, la intención de presentar la tabla 3 es mostrar las capacidades predictivas actuales de la teoría de KS, que se pueden resumir diciendo que: a) en propiedades energéticas, o más precisamente en diferencias de energía, las desviaciones son de aproximadamente 2-3kcal/mol y b) las distancias de enlace se predicen con una desviación respecto al experimento de aproximadamente 0.01-0.02Å. Probablemente quien no requiera de esta información no puede valorar las implicaciones que tienen estas pequeñas desviaciones, pero son lo suficientemente buenas como para poder predecir con confianza valores de estas propiedades en sistemas moleculares que sean muy diferentes de las moléculas que conforman los conjuntos de prueba. Incluso, cuando existe una discrepancia importante entre los valores predichos por la teoría y los determinados experimentalmente, el nivel de confianza permite sugerir que puede existir algún problema en la medición experimental.
Es posible mejorar de una manera sistemática y jerárquica la calidad de los resultados de los métodos de función de onda, empezando en el nivel más bajo, mediante la inclusión de excitaciones y la ampliación de la base. Este fue otro punto por el cual la DFT recibió y recibe un buen número de críticas. Para responder a estas críticas, el profesor John P. Perdew, una de las personalidades más importantes en el campo del desarrollo de funcionales de intercambio y de correlación, y quien es uno de los científicos más citados (de acuerdo con la base de datos Web of Science de Thomson Reuters, hasta julio de 2016, el trabajo del profesor Perdew ha recibido ¡más de 140,000 citas!), introdujo una propuesta para mejorar los resultados de la DFT, en su versión de KS, a la que bautizó como «escalera de Jacob» (Perdew y Schmidt, 2001), la cual es un camino jerárquico que, en principio, nos permite mejorar la calidad de las predicciones de la DFT de una manera sistemática. En la figura 1 mostramos nuestra versión azteca de la escalera de Perdew, donde se puede apreciar que conforme se suben los peldaños de la escalera, o la pirámide, las desviaciones respecto al experimento van disminuyendo hasta alcanzar, en la punta de la pirámide, la precisión que John Pople bautizó como el «cielo químico» y que corresponde a tener una metodología teórica capaz de predecir calores de formación con desviaciones menores a 1kcal/mol respecto al valor experimental. Los diferentes peldaños corresponden a las aproximaciones local (LDA), de gradientes generalizados (GGA), más allá de gradientes generalizados (meta-GGA), aún más allá de gradientes generalizados (hyper-GGA) y, finalmente, el escalón más alto en esta pirámide, la aproximación de fase aleatoria (RPA). Al subir por este camino se pueden mejorar los resultados de la DFT a expensas de una mayor demanda computacional. Hoy, muchos investigadores consideran la escalera de Jacob propuesta por Perdew como «el» camino para mejorar los resultados, pero hay que señalar que no es infalible, ni único y tampoco está libre de problemas. Mi cuestionamiento principal a la propuesta de Perdew es que a partir del nivel meta-GGA, la escalera de Jacob se aleja del espíritu original del primer teorema de Hohenberg y Kohn pues las aproximaciones no dependen exclusivamente de la densidad, sino que dependen de los orbitales de KS. Llegar al «cielo químico» sin recurrir a un lenguaje de orbitales es una de las fronteras de investigación de la DFT. A pesar de no estar libre de problemas, no tenemos muchas dudas sobre la enorme ventaja de usar la DFT como el método por elección para estudiar la estructura electrónica de la materia…, al menos en los años inmediatos por venir.
Perspectivas
El gran reto para estudiar la materia en condiciones humanas, esto es, a temperaturas y presiones donde existen seres humanos que lo puedan hacer, es que los sistemas de interés en Química tienen una multitud de escalas, tanto espaciales como temporales, hecho que se ilustra en la figura 2. Con este escalamiento transitamos desde el diseño molecular al diseño ingenieril, pasando por el diseño atomístico y mesoscópico. Los avances recientes en las teorías y algoritmos para estudiar la materia en múltiples escalas espaciotemporales nos permiten prever que en un futuro no muy lejano seremos capaces de diseñar objetos en escala humana, como coches, aviones, sistemas generadores y almacenadores de energía, catalizadores, fármacos selectivos, etc., partiendo desde la escala electrónica y llegando a la escala macroscópica, alcanzando, así, uno de los sueños de la Química Teórica: la comprensión del comportamiento de la materia desde primeros principios.
Conflicto de intereses
Los autores declaran no tener ningún conflicto de intereses.
La revisión por pares es responsabilidad de la Universidad Nacional Autónoma de México.