






Las hipolipoproteinemias se caracterizan por la disminución de la concentración plasmática de lipoproteínas. Dentro de estas, encontramos dos grupos: las hipobetalipoproteinemias (HBL), debidas a una disminución de la concentración plasmática de lipoproteínas que contienen apolipoproteína B, y las hipoalfalipoproteinemias.
Las hipolipoproteinemias pueden clasificarse en función de su origen, en primarias y secundarias. Las HBL primarias son entidades raras producidas por mutaciones en diferentes genes. Hasta ahora, se han identificado más de 140 mutaciones en los genes APOB, PCSK9, ANGPTL3, MTTP y SAR1. El diagnóstico y tratamiento precoz son fundamentales para evitar el desarrollo de complicaciones graves.
En la presente revisión abordamos el diagnóstico y tratamiento de las HBL, especialmente aquéllas que cursan con hipotrigliceridemia.
Hypolipoproteinemias are characterized by a decrease in the plasma concentration of lipoproteins. Within them, we find two groups: hypobetalipoproteinemias (HBL), due to a decrease in the plasma concentration of lipoproteins containing apolipoprotein B, and hypoalphalipoproteinemias.
Hypolipoproteinemias can be classified according to their origin, into primary and secondary. Primary HBLs are rare entities produced by mutations in different genes. So far, more than 140 mutations have been identified in the APOB, PCSK9, ANGPTL3, MTTP, and SAR1 genes. Early diagnosis and treatment are essential to avoid the development of serious complications.
In this review we address the diagnosis and treatment of HBL, especially those in which there is hypotriglyceridemia.
El concepto de dislipoproteinemia incluye tanto entidades que cursan con exceso, como con disminución o alteraciones de la composición de las lipoproteínas plasmáticas. En general, prestamos mayor atención a aquellas situaciones que se presentan con hiperlipidemia. Sin embargo, existen diferentes tipos de hipolipoproteinemias que pueden ocasionar complicaciones graves si no se tratan de forma adecuada.
Las hipolipoproteinemias se caracterizan por la disminución de la concentración plasmática de lipoproteínas. Dentro de estas, encontramos dos grupos: las hipobetalipoproteinemias (HBL), debidas a una disminución de la concentración plasmática de lipoproteínas que contienen apolipoproteína B (apoB), y las hipoalfalipoproteinemias. En la presente revisión nos centraremos en las primeras.
Las hipolipoproteinemias pueden clasificarse en función de su origen, en primarias y secundarias. Además, en función de la fracción alterada, se pueden clasificar en hipocolesterolemias, hipotrigliceridemias e hipolipidemias mixtas.
Las HBL primarias o familiares son poco frecuentes. Se clasifican en función del gen involucrado y del patrón de herencia, así como de la severidad de la mutación.
Las HBL secundarias son las más frecuentes. Diferentes situaciones, tales como el tipo de dieta o diversas patologías, pueden producir niveles reducidos de apoB. Las causas más frecuentes de HBL secundaria1 se recogen en la tabla 1.
Causas de hipobetalipoproteinemia secundaria
| 1. Desnutrición: dietas vegetarianas estrictas y alcoholismo crónico |
| 2. Malabsorción intestinal: pancreatitis crónica, enfermedad celíaca, fibrosis quística y acrodermatitis enterohepática |
| 3. Enfermedades hepáticas: síndrome de Reye, enfermedades del parénquima hepático y necrosis hepática |
| 4. Infecciones graves |
| 5. Infecciones crónicas |
| 6. Enfermedades que inducen estados inflamatorios crónicos |
| 7. Cáncer |
| 8. Tratamiento hipolipemiante |
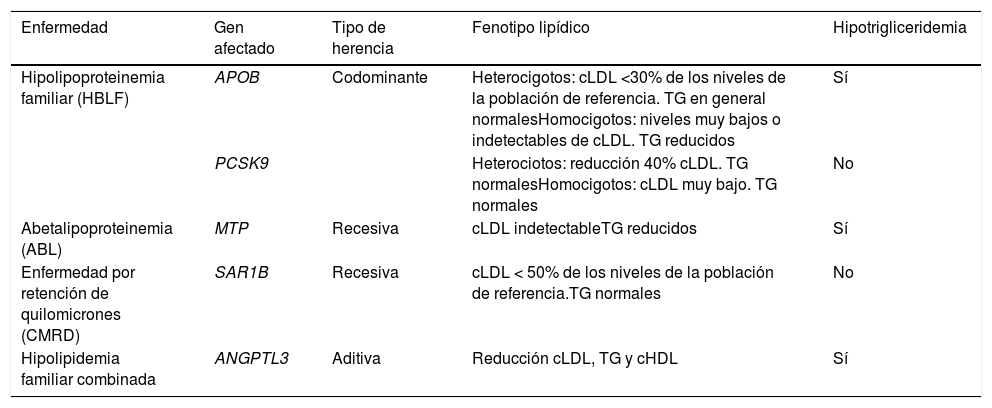
Las HBL primarias son entidades raras producidas por mutaciones en diferentes genes. Se pueden clasificar en función del fenotipo lipídico, el gen afectado y la forma de herencia, así como la gravedad de la mutación (tabla 2). El diagnóstico definitivo viene dado por el estudio genético. Hasta ahora, se han identificado más de 140 mutaciones en los genes APOB, PCSK9, ANGPTL3, MTTP y SAR1, de las cuales la mayoría son mutaciones puntuales que dan como resultado errores de empalme o truncamientos prematuros2–5.
Trastornos lipoproteicos que causan hipobetalipoproteinemia primaria
| Enfermedad | Gen afectado | Tipo de herencia | Fenotipo lipídico | Hipotrigliceridemia |
|---|---|---|---|---|
| Hipolipoproteinemia familiar (HBLF) | APOB | Codominante | Heterocigotos: cLDL <30% de los niveles de la población de referencia. TG en general normalesHomocigotos: niveles muy bajos o indetectables de cLDL. TG reducidos | Sí |
| PCSK9 | Heterociotos: reducción 40% cLDL. TG normalesHomocigotos: cLDL muy bajo. TG normales | No | ||
| Abetalipoproteinemia (ABL) | MTP | Recesiva | cLDL indetectableTG reducidos | Sí |
| Enfermedad por retención de quilomicrones (CMRD) | SAR1B | Recesiva | cLDL < 50% de los niveles de la población de referencia.TG normales | No |
| Hipolipidemia familiar combinada | ANGPTL3 | Aditiva | Reducción cLDL, TG y cHDL | Sí |
cHDL: colesterol HDL; cLDL: colesterol LDL; TG: triglicéridos.
La hipobetalipoproteinemia familiar (HBLF) se caracteriza por niveles plasmáticos de apoB por debajo del percentil 5 de la población de referencia, y por niveles de colesterol de las lipoproteínas de baja densidad (LDL) o colesterol LDL (cLDL) generalmente entre 20 y 50 mg/dL, así como por la posibilidad de niveles reducidos de triglicéridos (TG)6, en ausencia de causas secundarias. Aunque la prevalencia de HBLF es desconocida, se ha sugerido, con base en los resultados de estudios en la población de descendentes de Framingham, que podría estar en torno al 2%7. La HBLF o HBLPF? es consecuencia de múltiples mutaciones localizadas en los genes de APOB, o menos frecuentemente, de PCSK9 y angiopoietin-like proteins (ANGPTL3). Además, otros genes todavía no identificados deben estar también asociados con el desarrollo de HBLF2,8.
HBLF apoB específicaLa HBLPF apoB específica es un trastorno autosómico codominante que afecta a las lipoproteínas que contienen apoB (HBLF1, OMIM 615558). Su prevalencia se estima en torno a uno de cada 1.000 a 3.000 individuos. Se han descrito más de 60 mutaciones en el gen de APOB que producen la HBLF19. Este gen se localiza en el cromosoma 2 y consta de 29 exones. Codifica dos proteínas, la apoB-100, de origen hepático y la apoB-48, de origen intestinal. ApoB es el componente principal de los quilomicrones (apoB-48), lipoproteínas de muy baja densidad (VLDL) (apoB-100) y LDL (apoB-100)10. Estas mutaciones anulan o interfieren la traducción de apoB y, generalmente, dan lugar a una proteína apoB truncada de diferente longitud. Como consecuencia, se produce una alteración de la síntesis y la secreción de VLDL por el hígado, así como el incremento del catabolismo de las lipoproteínas que contienen apoB (VLDL y LDL)11.
El fenotipo clínico depende del número de alelos afectados. Los pacientes con HBLF pueden ser homocigotos o heterocigotos. Los individuos heterocigotos generalmente son asintomáticos, y son identificados por la presencia de niveles reducidos de lípidos. Estas personas tienen menor riesgo de enfermedad cardiovascular que la población general. Sin embargo, un porcentaje considerable de ellas puede desarrollar esteatosis hepática, como consecuencia del acúmulo de TG en los hepatocitos, debido a la alteración de la secreción de VLDL11,12. Incluso en algunos casos, si bien es poco frecuente, puede haber progresión de la enfermedad hepática, conduciendo al desarrollo de cirrosis hepática y carcinoma hepatocelular, especialmente cuando existe asociación con otros factores de riesgo13. La severidad de la enfermedad hepática depende de la capacidad de la apoB truncada para ser «lipidada». Asimismo, en ocasiones pueden presentar ligera malabsorción de grasa.
Por el contrario, la forma homocigota y la heterocigota compuesta pueden presentar manifestaciones clínicas importantes si no son tratadas de forma adecuada. La sintomatología es similar a la observada en la abetalipoproteinemia (ABL).
El diagnóstico de la HBLF1 es eminentemente clínico. Se basa en la presencia de concentraciones de colesterol total, cLDL y apoB por debajo del percentil 5 de la población de referencia. Asimismo, los pacientes suelen presentar niveles plasmáticos de TG disminuidos, especialmente en el caso de las formas homocigotas.
El diagnóstico diferencial preliminar entre pacientes homocigóticos y heterocigóticos se basa en una evaluación de los niveles de lípidos. En individuos homocigotos, los niveles de colesterol total y cLDL que confirman el diagnóstico son < 80 y < 20 mg/dL, respectivamente, mientras que en los heterocigotos los niveles plasmáticos de colesterol total y cLDL suelen ser < 120 y < 80 mg/dL, respectivamente. En la forma homocigótica de HBLF1, el nivel de TG es muy bajo, mientras que en la forma heterocigótica de esta enfermedad, suele ser normal13.
El estudio familiar es muy importante. Debe mostrar una herencia autosómica dominante (a diferencia de la ABL). Además, se deben descartar causas secundarias de HBL. El diagnóstico definitivo vendrá dado por el análisis de mutaciones en el gen de apoB2,8,11.
Muchos de los pacientes con HBLF1 presentan incremento de transaminasas debido a la esteatosis hepática y, en ocasiones, una ligera malabsorción de grasa. Dados los hallazgos recientes del desarrollo de cirrosis y carcinoma hepatocelular asociados con HBLF1, es aconsejable monitorizar las enzimas hepáticas, y en caso de estar elevadas, considerar la realización de pruebas de imagen13. Además, se debe evaluar la presencia de deficiencia de vitaminas liposolubles.
Los trastornos gastrointestinales responden satisfactoriamente a la restricción de TG de cadena larga. Además, se debe suplementar los ácidos grasos esenciales. Asimismo, los pacientes con HBLF pueden presentar déficit de vitaminas liposolubles, siendo necesaria su suplementación. Con respecto a los heterocigotos, la restricción de grasa en la dieta solo se recomienda en caso de existir malabsorción. Sí podría estar indicado el suplemento con vitamina E en cantidades moderadas para prevenir el desarrollo de afectación neurológica12.
HBLF por pérdida de función de PCSK9La HBLF también puede deberse a la presencia de mutaciones en el gen de PCSK9, que codifica una proteasa que se une al receptor de LDL, induciendo la degradación lisosomal hepatocitaria12. Como consecuencia de mutaciones con pérdida de función en este gen, se incrementa el número de receptores de LDL en la superficie celular, y por tanto, se reducen los niveles plasmáticos de cLDL y de apoB, lo que conduce a una disminución del riesgo de desarrollar enfermedad cardiovascular. Los niveles de triglicéridos no están reducidos. Estos pacientes no presentan ninguna de las manifestaciones clínicas asociadas con las HBLF dependientes de la apoB2,12,14. Se trata de un trastorno benigno que no requiere tratamiento.
AbetalipoproteinemiaLa ABL (OMIM #200100), también conocida como síndrome de Bassen-Kornzweig, es una hipolipidemia autosómica recesiva muy rara (< un caso por cada millón de habitantes) caracterizada por la virtual ausencia de las lipoproteínas plasmáticas que contienen apoB1,12. La ABL es consecuencia de mutaciones en ambos alelos del gen proteína transferidora microsomal de triglicéridos (MTP) en el cromosoma 4. La MTP es una proteína crucial en la formación de las lipoproteínas ricas en TG. Cataliza la transferencia de TG a la partícula de apoB naciente en el ensamblaje de TG de VLDL en el retículo endoplásmico rugoso, mientras la apoB se transloca cotranslacionalmente a través de la membrana del retículo endoplásmico15,16.
La clínica de la ABL afecta a múltiples órganos y es generalmente indistinguible de la clínica de la HBLF1 homocigota y de la HBLF1 en heterocigotos compuestos, y mucho más grave de la que presentan los sujetos con HBLF1 heterocigotos. Sin embargo, la presentación clínica puede ser heterogénea, dependiendo sus complicaciones posteriores de la rapidez del diagnóstico y del inicio del tratamiento.
El principal hallazgo clínico de la ABL es la presencia de síndrome de malabsorción grasa. Suele observarse ya en el período neonatal con esteatorrea, vómitos, distensión abdominal y retraso del crecimiento. Esta sintomatología disminuye de forma considerable cuando el paciente ingiere una dieta baja en grasas. Otros síntomas pueden involucrar a diferentes órganos como consecuencia de la deficiencia de ácidos grasos esenciales y de vitaminas liposolubles. Así, los individuos pueden desarrollar disartria, arreflexia, retinitis pigmentaria y ataxia grave, lo que conduce a la reducción de la esperanza de vida de forma considerable. Asimismo, pueden desarrollar enfermedad hepática. Además, pueden presentar alteraciones hematológicas, como acantocitosis, anemia y hemólisis, entre otras17,18.
En la ABL, se puede hacer un diagnóstico clínico basado en el perfil lipídico, el frotis sanguíneo y la presencia de síntomas y signos característicos. Como se ha comentado previamente, todos los pacientes con ABL tienen esteatorrea debido a malabsorción de grasas. Dependiendo de la edad de la primera manifestación, la mayoría de los sujetos también presentan anomalías neurológicas, en mayor o menor grado, debido a la profunda deficiencia de vitamina E. Además, la presencia de síntomas que involucran a otros órganos se pueden utilizar para respaldar la sospecha clínica. Entre las determinaciones de laboratorio, el frotis sanguíneo muestra la presencia de acantocitosis, y el perfil lipídico revelaría niveles plasmáticos casi indetectables de cLDL, TG y apoB17. El diagnóstico diferencial se debe realizar fundamentalmente con tres entidades: la HBLF1 homocigota, cuya sintomatología es indistinguible de la ABL, pero que tiene patrón de herencia autosómico dominante; con el síndrome de neuroacantocitosis de McLeod, que cursa con acantocitosis y neuropatía, pero sin alteraciones lipídicas ni manifestaciones debidas a la deficiencia de vitaminas liposolubles; y con la ataxia de Friedrich, que se presenta con síntomas neurológicos similares, pero sin alteraciones lipídicas ni manifestaciones provocadas por la deficiencia de vitaminas liposolubles18.
Además, muchos de los pacientes con ABL presentan incremento de transaminasas debido a esteatosis hepática, siendo aconsejable monitorizar los enzimas hepáticos y considerar la realización de pruebas de imagen13. También se debe evaluar la presencia de deficiencia de vitaminas liposolubles.
El diagnóstico definitivo vendrá dado por el análisis de mutaciones en el gen de MTP12,16,17. Se han descrito más de 30 mutaciones causales que producen ABL.
Respecto al seguimiento, Lee et al. recomiendan una evaluación clínica cada seis a 12 meses, incluida la valoración de la dieta y cualquier síntoma gastrointestinal o neurológico. Además, sugieren la determinación analítica del perfil lipídico, la función hepática y el estudio hematológico de forma anual. Asimismo, es recomendable la realización de una ecografía hepática cada tres años17.
En lo que respecta al manejo de la ABL, el tratamiento estándar es la restricción de grasas en la dieta (< 30% del total de calorías) que puede eliminar la esteatorrea, junto con suplementos de vitaminas liposolubles. Se recomienda la administración oral de suplementos de vitamina E en dosis altas (100 a 300 mg/kg/día) para detener la progresión de la enfermedad neurológica. Además, la suplementación con una combinación de altas dosis de vitaminas E y A es eficaz para reducir la degeneración de la retina17.
Enfermedad de retención de quilomicronesLa enfermedad de retención de quilomicrones (CMRD, OMIM #246700), también conocida como enfermedad de Anderson, es una patología muy rara (menos de un caso por millón de personas) autosómica recesiva caracterizada por la incapacidad de los enterocitos para producir quilomicrones, con el consiguiente acúmulo de lípidos (en forma de pre-quilomicrones) en el citoplasma de estas células2.
La CMRD está causada por mutaciones en el gen SAR1B (o SARA2), que codifica la proteína Sar1B, involucrada en el transporte de quilomicrones desde el retículo endoplásmico hasta el aparato de Golgi. La presencia de esta mutación impide la fusión de la vesícula transportadora de pre-quilomicrones con el aparato de Golgi, lo que conduce a un acúmulo de estas vesículas en el citoplasma de los enterocitos. Además, Sar1B también participa en la secreción hepática de VLDL, por lo que también existe una acumulación de lípidos en los hepatocitos19.
La clínica aparece poco después del nacimiento y se caracteriza por diarrea, malabsorción de grasa, retraso en el crecimiento y vómitos con distensión abdominal. Al igual que en otros tipos de hipolipidemia familiar, puede existir deficiencia de vitaminas liposolubles y esteatosis hepática, aunque no se ha descrito la evolución hacia formas graves. Asimismo, la afectación neurológica y oftalmológica también es menos grave que en otros tipos de hipolipidemia familiar.
El diagnóstico generalmente se realiza con retraso debido a la sintomatología poco específica. Se basa en la presencia de diarrea crónica con malabsorción de grasa y un perfil lipídico anormal, así como el retraso en el crecimiento. A nivel bioquímico, se objetiva la presencia de niveles plasmáticos de colesterol disminuidos y aumento de transaminasas.
No hay recomendaciones específicas para el tratamiento y el seguimiento, por lo que, en la actualidad, consideran lo sugerido para la ABL. La terapia temprana puede prevenir la aparición de la mayoría de las secuelas. Esta incluye restricción de TG de cadena larga, además de suplementación importante de vitamina E y de otras vitaminas liposolubles, así como de ácidos grasos esenciales1,12,20.
Hipolipidemia familiar combinadaLa hipolipidemia familiar combinada, también conocida como HBLF2 (OMIM #605019), es un trastorno hereditario caracterizado por niveles plasmáticos extremadamente bajos de cLDL, colesterol de las lipoproteínas de alta densidad (HDL) o colesterol HDL (cHDL) y TG21.
El patrón de transmisión de esta entidad es complejo. Si bien, inicialmente fue considerado como un trastorno recesivo, los estudios de segregación familiar sugieren que el modo de herencia es aditivo22,23.
Se debe a mutaciones con pérdida de función en el gen angiopoietin-like 3 (ANGPTL3) que codifica la proteína ANGPTL3, secretada casi exclusivamente por el hígado. Esta ejerce un importante efecto regulador de los niveles plasmáticos de TG y colesterol, mediante inhibición reversible de las actividades enzimáticas de la lipoproteina lipasa y de la lipasa endotelial5.
Clínicamente, se caracteriza por la presencia de panhipolipidemia, con niveles plasmáticos de cLDL, cHDL y TG extremadamente bajos. A diferencia de la HBLF1, en la HBLF2 no existe un incremento de la prevalencia de enfermedad hepática crónica respecto a la población general11. Los pacientes con HBLF2 presentan una reducción significativa del riesgo de enfermedad cardiovascular24.
El diagnóstico se basa en los hallazgos de laboratorio previamente descritos, así como en la detección de mutaciones en el gen ANGPTL3 con pérdida de función. Los individuos con esta entidad no requieren tratamiento.
ConclusiónLas hipolipidemias familiares son patologías muy poco frecuentes, pero que pueden tener consecuencias graves en caso de no ser diagnosticadas y tratadas de forma precoz. La sospecha clínica viene dada por el hallazgo analítico de cifras de lípidos reducidas, asociadas o no a sintomatología variable. El diagnóstico definitivo lo proporcionará el estudio genético.
FinanciaciónSergio Martinez-Hervas es un investigador del programa Juan Rodes (JR18/00051) financiado por el Instituto de Salud Carlos III y el Fondo Europeo para el Desarrollo Regional (FEDER).
Conflicto de interesesEl Dr. Sergio Martínez-Hervás y el Dr. José Tomás Real-Collado declaran que este artículo forma parte de una monografía que ha sido patrocinada por Akcea.
El Dr. Juan Francisco Ascaso-Gimilio declara que este artículo forma parte de una monografía que ha sido patrocinada por Akcea y que colabora con Mylan en el grupo de dislipemia aterogénica de la SEA, con Ferrer en el grupo de hipertrigliceridemias y con MSD en un curso anual de dislipemia y diabetes, no relacionados con la realización del trabajo.
Nota al suplementoEste artículo forma parte del suplemento «Diagnóstico y tratamiento de las alteraciones del metabolismo de los triglicéridos: de la fisiopatología a la práctica clínica», que cuenta con el patrocinio de Akcea Therapeutics.
Al CIBER de Diabetes y Enfermedades Metabólicas Asociadas (CIBERDEM) que es una iniciativa del Instituto de Salud Carlos III.