








“Kernicterus” is a term currently used to describe bilirubin induced brain injury in the neuro-pathological studies. This is a confusing term and nowadays we prefer bilirubin encephalopathy or bilirubin induced neurological dysfunction. The clinical signs vary and it is clearly decreasing in prevalence in developed countries.
Materials and methodsWe review a series of 7 patients with bilirubin encephalopathy and variable neurological manifestations, who were seen in the Neuropaediatric Department in the last 10 years. Only one patient died in the neonatal period with hyperbilirubinaemia, sepsis and multi-organ failure.
ResultsDiverse aetiological factors were related to hyperbilirubinaemia. All patients had clinical symptoms due to hyperbilirubinaemia. Neuroimaging during the neonatal period showed involvement of the nucleus pallidus, with hyperintensity in T1 in the brain MR scan as the most consistent finding. All the patients who survived developed neurological signs and we try to correlate them with biochemical, clinical, neuroimaging and neurophysiological parameters.
ConclusionsAn increase in the number of patients with bilirubin encephalopathy has been observed over the last few years, and we attempt to find out the causes. The increased survival of the low birth weight newborns, the increase in the immigration population and the use of diagnostic neuroimaging contribute to this increase. It is a great challenge for the neonatologist and for neuropaediatricians to prevent its occurrence and to minimise the effects of bilirubin encephalopathy.
“kernicterus” se aplicó inicialmente a la tinción amarilla de los ganglios basales en estudios necrópsicos, pero es un término impreciso y se habla más de encefalopatía por bilirrubina o de disfunción neurológica inducida por la bilirrubina. Clínicamente la toxicidad por hiperbilirrubinemia puede ser muy variable y en países desarrollados tiende a desaparecer.
Material y métodosRevisamos una serie de 7 pacientes con encefalopatía por bilirrubina y diferentes grados de compromiso neurológico, atendidos en los últimos 10 años en el Servicio. Solo falleció un paciente en período neonatal con hiperbilirrubinemia, sepsis y fallo multiorgánico.
ResultadosLas causas etiológicas de la hiperbilirrubinemia fueron muy variadas. Los 7 pacientes presentaron ictericia, clínica neonatal, la neuroimagen ya permitió demostrar las lesiones en núcleo pálido con hiperintensidad de T1. Todos los pacientes presentaron manifestaciones clínicas en período neonatal, y secuelas neurológicas más o menos graves en los 6 supervivientes que se intentan correlacionar con los demás parámetros bioquímicos, clínicos, de neuroimagen y neurofisiológicos.
ConclusionesHemosconstatadounincrementodelasobservacionesdedisfunciónneurológica inducida por la bilirrubina y nos planteamos conocer las causas de esta situación. La mayor supervivencia de los grandes prematuros, el aumento de la población inmigrante y la posibilidad del diagnóstico por neuroimagen contribuyen a este incremento. Continua siendo un reto para el neonatólogo y el neuropediatra evitar su presentación y minimizar los efectos de la toxicidad por bilirrubina en período neonatal.
The term “kernicterus” was originally used to denote the yellow staining of the basal ganglia in pathological samples from patients who died with jaundice due to erythroblastosis fetalis.1 The term was subsequently applied to static encephalopathy with dystonic tetraparesis and hearing loss produced by hyperbilirubinaemia during the neonatal period.1,2
Other authors recommend using “kernicterus” to designate any manifestation arising from neonatal hyperbilirubinaemia, with clinical signs ranging from mild sensorineural hearing loss to more severe cases of intellectual disability, deafness or choreoathetosis.2
Currently, we prefer the terms “bilirubin encephalopathy” or “bilirubin-induced neurological dysfunction” when referring to all of the different manifestations of untreated or insufficiently treated neonatal hyperbilirubinaemia.
Bilirubin – a breakdown product of haemoglobin catabolism – is a pigment which is extremely toxic to biological systems, and especially to the nervous system.1,2 Unconjugated bilirubin enters the neuron as the result of biochemical processes that are not fully understood, but which are accepted as probable mechanisms. Brodersen3 and Wennberg4 proposed a model according to which bilirubin can adapt to the neuron cell membrane by means of a delicate balance involving the following factors (and others): serum albumin level, pH of the medium, and changes in the internal structure of the bilirubin molecule. Pigment may enter and exit the neuron cytoplasm depending on the stability of the factors listed above.3,5 Experiments have shown that bilirubin bound to albumin can pass through a blood-brain barrier (BBB) that is damaged by use of hypertonic agents. Within the neuron, bilirubin disrupts the proton gradient and directly interferes in intramitochondrial oxidative processes, finally eliciting apoptosis and neuronal necrosis.2–4
The introduction of magnetic resonance imaging (MRI) in daily clinical practice in neonatal units let us view the kernicterus lesions which could previously only be seen through anatomical pathology studies. Several excellent recent studies address this matter.6,7 During the acute phase, these changes are probably indicative of a gemistocytic reaction by astrocytes. Once the acute phase is over, if MR images are still abnormal, these findings probably reflect the dense fibrillar gliosis with a low cellular content that appears in the final phase.8
At present, we are observing an increase in the number of patients with bilirubin encephalopathy due to several reasons which we will analyse shortly. For that reason, we believe that presenting our experience and literature review is relevant.
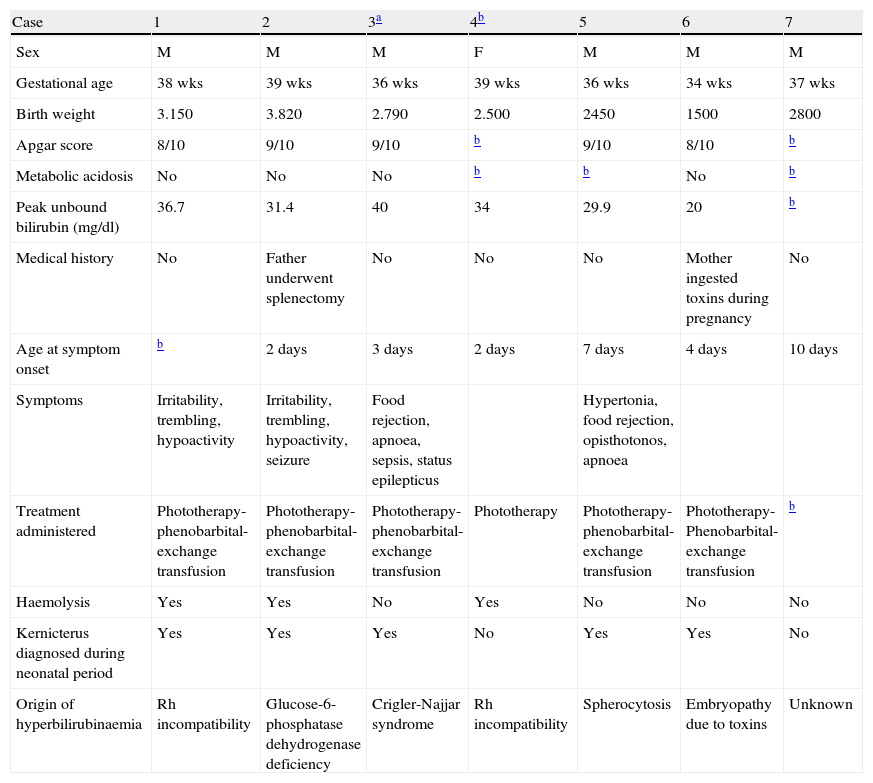
Patients and methodsWe reviewed medical histories and neuroimaging studies (MRIs and transfontanelle cranial ultrasound) from 7 patients who received care in the hospital's neurology department between 1999 and 2008. These images allowed us to identify neurological sequelae stemming from neonatal hyperbilirubinaemia (Table 1). Males accounted for 6 of the 7 patients, none of the 7 was born in our hospital, and 2 were not born in Spain. Some were referred during the neonatal period, and others were older when they were first examined. We analysed brain tissue samples obtained from the autopsy of the patient who died at our hospital. In this series, only 2 patients were not diagnosed with bilirubin encephalopathy as neonates. Haemolysis was found in 3 cases, and the origins of hyperbilirubinaemia were very diverse (Rh incompatibility in 2 cases; 1 case each of glucose-6-phosphatase dehydrogenase deficiency, Criglar Najjar syndrome, spherocytosis, and embryopathy due to toxins; and 1 of unknown causes in the case of a patient born outside of Spain).
Summary of clinical and biochemical data from patients with kernicterus (HSJD), 1998–2008.
| Case | 1 | 2 | 3a | 4b | 5 | 6 | 7 |
| Sex | M | M | M | F | M | M | M |
| Gestational age | 38 wks | 39 wks | 36 wks | 39 wks | 36 wks | 34 wks | 37 wks |
| Birth weight | 3.150 | 3.820 | 2.790 | 2.500 | 2450 | 1500 | 2800 |
| Apgar score | 8/10 | 9/10 | 9/10 | b | 9/10 | 8/10 | b |
| Metabolic acidosis | No | No | No | b | b | No | b |
| Peak unbound bilirubin (mg/dl) | 36.7 | 31.4 | 40 | 34 | 29.9 | 20 | b |
| Medical history | No | Father underwent splenectomy | No | No | No | Mother ingested toxins during pregnancy | No |
| Age at symptom onset | b | 2 days | 3 days | 2 days | 7 days | 4 days | 10 days |
| Symptoms | Irritability, trembling, hypoactivity | Irritability, trembling, hypoactivity, seizure | Food rejection, apnoea, sepsis, status epilepticus | Hypertonia, food rejection, opisthotonos, apnoea | |||
| Treatment administered | Phototherapy-phenobarbital-exchange transfusion | Phototherapy-phenobarbital-exchange transfusion | Phototherapy-phenobarbital-exchange transfusion | Phototherapy | Phototherapy-phenobarbital-exchange transfusion | Phototherapy-Phenobarbital-exchange transfusion | b |
| Haemolysis | Yes | Yes | No | Yes | No | No | No |
| Kernicterus diagnosed during neonatal period | Yes | Yes | Yes | No | Yes | Yes | No |
| Origin of hyperbilirubinaemia | Rh incompatibility | Glucose-6-phosphatase dehydrogenase deficiency | Crigler-Najjar syndrome | Rh incompatibility | Spherocytosis | Embryopathy due to toxins | Unknown |
Table 1 presents a summary of data from all 7 patients. Only 1 patient was born pre-term (34 weeks); all others were full-term with normal birth weights according to gestational age. None of the patients experienced difficulties during birth, and their Apgar scores were normal. There was no evidence of metabolic acidosis. We noted 1 case in which the patient's father had undergone splenectomy and a second case in which the patient's mother used drugs during pregnancy. In all cases, clinical symptoms were present from the first week of life, especially during the first 72h after birth; patients showed jaundice, irritability, trembling, hypoactivity and abnormal movements. Peak unconjugated bilirubin in these patients was within the range of 20–40mg/dl between days 2 and 10 of life. All patients were treated with phenobarbital, intensive phototherapy and exchange transfusion, with varying results. Bilirubin levels normalised within 6 to 10 days after birth.
All the patients studied presented abnormal EEG findings of different types. During the neonatal period, 3 patients suffered epileptic seizures and 1 died in the neonatal ICU with sepsis, status epilepticus, multiple organ dysfunction and coma. The other patients presented sensorineural hearing loss and spastic-dystonic tetraparesis as neurological sequelae. Bilirubin encephalopathy was diagnosed during the first 18 days of the neonatal period in 5 cases, and during monitoring at a later point in the remaining cases.
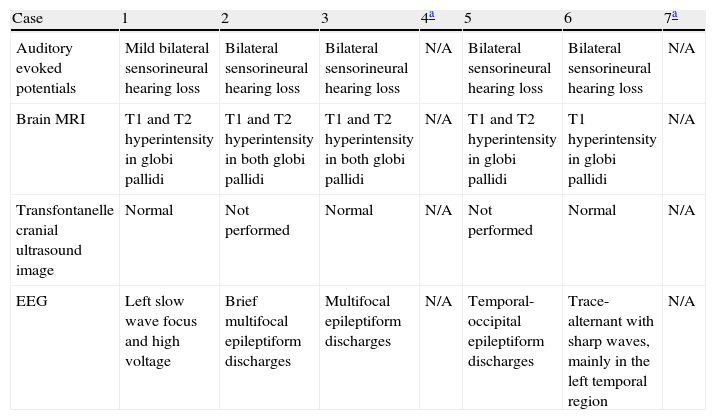
Neuroimaging findingsTransfontanelle cranial ultrasound images were normal in all studies. Cranial MRI was taken between days 6 and 17 of life. In all patients, we observed bilateral signal increases in both globi pallidi in T1-weighted sequences. In 3 patients, we also observed signal increases in the nuclei pallidi in T2-weighted sequences (Table 2) (Figs. 1–3).
Complementary study of patients with kernicterus during neonatal period/first months of life.
| Case | 1 | 2 | 3 | 4a | 5 | 6 | 7a |
| Auditory evoked potentials | Mild bilateral sensorineural hearing loss | Bilateral sensorineural hearing loss | Bilateral sensorineural hearing loss | N/A | Bilateral sensorineural hearing loss | Bilateral sensorineural hearing loss | N/A |
| Brain MRI | T1 and T2 hyperintensity in globi pallidi | T1 and T2 hyperintensity in both globi pallidi | T1 and T2 hyperintensity in both globi pallidi | N/A | T1 and T2 hyperintensity in globi pallidi | T1 hyperintensity in globi pallidi | N/A |
| Transfontanelle cranial ultrasound image | Normal | Not performed | Normal | N/A | Not performed | Normal | N/A |
| EEG | Left slow wave focus and high voltage | Brief multifocal epileptiform discharges | Multifocal epileptiform discharges | N/A | Temporal-occipital epileptiform discharges | Trace-alternant with sharp waves, mainly in the left temporal region | N/A |
N/A: data not available.
. Clear hyperintensity in the globus pallidus and faint hyperintensity in subthalamic nucleus (Case 5).")
.")
.")
Autopsy was performed on the patient who died in order to establish the aetiological diagnosis of the syndrome. On the macroscopic level, brain weight was 410 grams, and the structure showed no abnormalities in its folds or at the base of the skull (no signs of uncal herniation). Yellow staining was observed on the left cerebral peduncle, putamen, globi pallidi and substantia nigra in both cerebral hemispheres, as well as in the anterior perimedial area of both cerebellar hemispheres (Fig. 4).
. T2 FLAIR coronal MRI of the same case.")
On the microscopic level, we observed cerebral abnormalities compatible with hypoxic–ischaemic brain injury (oedema, neuronal retraction and nuclear picnosis associated with astrocytosis) in superficial cortical layers. In deep brain structures (putamen, globus pallidus, subthalamic nucleus, thalamus and hypothalamus and cerebral peduncles), we observed degenerative neuronal abnormalities including loss of Nissl substance, eosinophilic degeneration, nuclear vacuolation and necrosis with loss of neurons. These processes were accompanied by astrocytosis and occasional axonal spheroids (Fig. 5).
 (Case 3). Degenerative neuronal changes showing neuronal loss and astrocytosis.")
On the cerebellar level, we found ochre pigmentation in the cytoplasm of some Purkinje cells, with degeneration and focal loss of these cells together with moderate astrocytosis. The internal granular layer was preserved.
The immunohistochemical study of hepatocytes revealed a total lack of bilirubin UDP-glucuronosyltransferase enzyme, which was compatible with Type I Crigler-Najjar syndrome. However, we did not identify any mutations in the UGT1A1 gene.
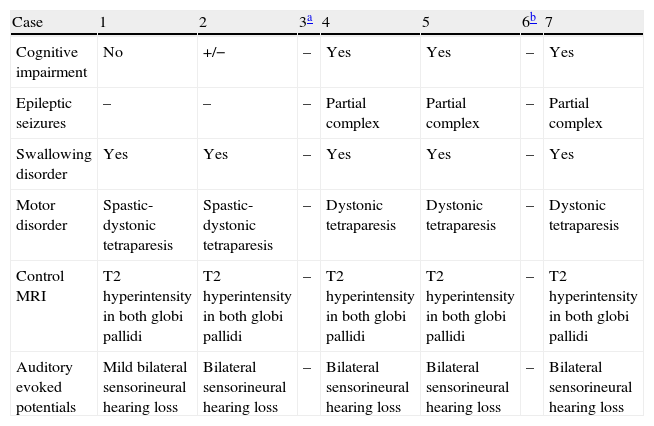
Course of the diseaseOf the 6 surviving patients, all demonstrate different degrees of neurological impairment (6/6), manifesting as spastic-dystonic tetraparesis in 2 cases and dystonic tetraparesis in 3 cases, with sensorineural hearing loss in 4 of the above 5 cases, epilepsy in 3 of the 5, and trouble swallowing in 5 of 5. Cognitive impairment was present in 3 of those 5 patients; only 1 patient is able to attend regular school with the aid of special materials. In all these patients, the syndrome manifested as static encephalopathy, and epilepsy is generally controlled by AEDs (Table 3).
Clinical course and complementary tests during monitoring of patients affected by kernicterus.
| Case | 1 | 2 | 3a | 4 | 5 | 6b | 7 |
| Cognitive impairment | No | +/− | – | Yes | Yes | – | Yes |
| Epileptic seizures | – | – | – | Partial complex | Partial complex | – | Partial complex |
| Swallowing disorder | Yes | Yes | – | Yes | Yes | – | Yes |
| Motor disorder | Spastic-dystonic tetraparesis | Spastic-dystonic tetraparesis | – | Dystonic tetraparesis | Dystonic tetraparesis | – | Dystonic tetraparesis |
| Control MRI | T2 hyperintensity in both globi pallidi | T2 hyperintensity in both globi pallidi | – | T2 hyperintensity in both globi pallidi | T2 hyperintensity in both globi pallidi | – | T2 hyperintensity in both globi pallidi |
| Auditory evoked potentials | Mild bilateral sensorineural hearing loss | Bilateral sensorineural hearing loss | – | Bilateral sensorineural hearing loss | Bilateral sensorineural hearing loss | – | Bilateral sensorineural hearing loss |
While the term “kernicterus”, referring to the yellowish tint of basal ganglia which can only be seen in autopsy studies, has been in use for many years, it is currently considered imprecise and the preferred term is bilirubin encephalopathy.2,3,9 This term is a useful one, as it indicates that different areas of the central nervous system may be affected, and reminds us that yellow staining does not have to appear in order for clinically recognisable neuronal dysfunction to be present. Macroscopic abnormalities in basal ganglia due to bilirubin encephalopathy can currently be detected by means of cranial MRI.1,2,6,7
It is now accepted that serum bilirubin levels higher than 20mg/dL increase the risk of neurological damage in full-term neonates, and we also know that pre-term neonates may suffer significant sequelae at much lower levels. This is especially true in the presence of additional factors such as hypoxia, acidosis, sepsis, haemolysis, polyglobulia or BBB disruption.4,5 When a BBB disruption occurs, bilirubin bound to albumin rapidly penetrates the cerebral extracellular space, while unconjugated bilirubin elicits severe neurotoxicity. Within the neuron, bilirubin disrupts the proton gradient, interferes with calcium homeostasis and creates neuronal hyperexcitability through excitatory amino acids and a neurotransmitter imbalance. It also exerts negative effects on the neuronal membrane, and lastly, it interferes directly with intramitochondrial oxidative processes and energy production, leading to neuronal apoptosis and necrosis.2–4 Other factors to be taken into account in the genesis of neuronal damage are the period of exposure to bilirubin, which varies with gestational age, or length of exposure to high bilirubin levels.4,5,9
It was recently suggested that when the BBB is intact, it uses an ATP-dependent transport system which acts like an efflux pump to remove unconjugated bilirubin, and maintains the concentration gradient of unconjugated bilirubin between plasma and the CSF.10 There are other factors that contribute to the increase of bilirubin, including the presence of substances that displace it from its albumin binding sites (sulphonamides) or which compete for albumin binding sites. The presence of sepsis, haemolysis, metabolic acidosis and especially pre-term birth are factors that contribute to developing bilirubin toxicity.3,10
The term “bilirubin encephalopathy” defines a neurological syndrome that arising when unconjugated bilirubin is deposited on certain cerebral cells. This phenomenon gives rise to yellowish staining and the destruction of the affected neurons. Several areas are more affected by this phenomenon than others, including the globi pallidi, subthalamic nuclei and hippocampus. Areas affected to a lesser extent are the thalamus, substantia nigra, cerebellar nuclei and certain nuclei in cranial nerve pairs. The cortex, the white matter and the brainstem are generally not affected.4,7,8,11
Clinically, hyperbilirubinaemia toxicity may be reversible and elicit no manifestations, or only late-onset, very subtle manifestations such as attention or auditory deficits or minimal motor impairment.2,5 In other cases, neurological symptoms may be more or less florid. Manifestations of bilirubin encephalopathy may be categorised as acute, chronic, or subtle, as described above.2
During the neonatal period, the classic clinical profile of bilirubin encephalopathy manifests with largely non-specific symptoms. These include feeding problems, irritability, depressed sensory cortex, convulsions or changes in muscle tone (hypertonia and hypotonia, retrocollis, opisthotonos), high-pitched (cerebral) cry, upward gaze, fever, and coma which may precede death. In our series of 7 patients, each one developed neurological symptoms in the neonatal period which were associated with different systemic manifestations reflecting their precarious state of health. During this time frame, BAEPs showed elongation of I–III waves and I–V waves, decreased amplitude of III and V waves, and even suppression of responses. Compatible BAEP abnormalities were detected in the 5 patients studied during the neonatal period or during the first months of life while under neonatal care. Improvement in BAEP activity was reported after exchange transfusion.5,12–14 Some authors have proposed establishing a prognostic score for acute-phase bilirubin encephalopathy.2,12
During this acute phase, transfontanelle cranial ultrasound imaging may show basal ganglia anomalies (which we were unable to detect in our cases), and in particular, head MRI images may show signal abnormalities in T1-weighted sequences in the globus pallidus or even the subthalamic nucleus.6 In all patients in our series with an available head MRI from the neonatal period, we observed an increase in the T1-weighted signal in globi pallidi and in one case, the subthalamic nuclei (Fig. 1). In 3 patients, hyperintensity was also reflected in the T2-weighted signal (Figs. 2 and 3).
The chronic form of the illness is characterised by a classic triad of motor, auditory, and oculomotor deficits. Motor manifestations correlate perfectly with the topographical location of bilirubin penetration in basal ganglia (nucleus pallidus, subthalamic nucleus, cerebellum and brain stem). Bilirubin penetration in the nucleus of cranial nerve pair VIII and the auditory nerve correlates with sensory deficit. Lastly, damage to the nuclei of the oculomotor nerves near the brain stem explains the abnormal eye movements typical in bilirubin encephalopathy.2,11,12 Patients may also show associated cognitive impairment, epilepsy, microcephaly and dental enamel abnormalities.2
The most important sequela is motor impairment with dystonic or spastic-dystonic tetraparesis. The symptom generally manifests as static encephalopathy with an overall poor response to physiotherapy, stimulation and medications (Table 3).2,12,15 Only one patient in our series was able to walk unaided by the age of 4 years.
Neurosensory deficit is a common, well-known manifestation of bilirubin toxicity.4,10,12–14 However, Starr et al in 1996 described an auditory neuropathy, also known as auditory dyssynchrony, characterised by lack of response to BAEP with preserved otoacoustic emissions and a variable degree of hearing loss.13 These patients experience difficulties understanding language, even in the absence of significant hearing loss, language delay, behavioural disorders and learning disabilities. This syndrome is present in between 5.3% and 14.8% of neonates cared for in an ICU, and it is more common where there is a history of pre-term birth and hyperbilirubinaemia.13 Patients gain scant benefits from hearing aid use, but results with cochlear implants are more encouraging.16 Oculomotor signs (impairment of upward gaze and, more rarely, impairment of horizontal eye movements similar to oculomotor apraxia) are not always easy to detect in the neonatal period, and tend to improve over time.2 Our series reports anomalies in eye movements in 4 patients during the neonatal period.
Bilirubin penetration tends not to occur in the cerebral cortex and subcortical white matter, so patients do not always suffer cognitive impairment. However, in our series, more than half of the patients had cognitive sequelae. In other series, rates of intellectual disability and epilepsy may be quite high.2,8
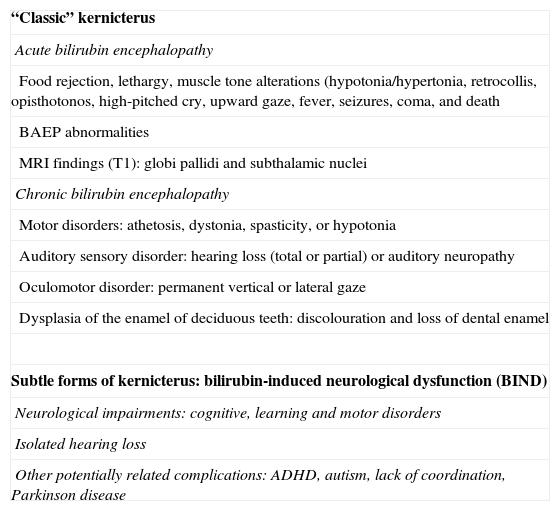
At present, the historical term “kernicterus” is losing favour. It is now thought to refer to an entire clinical spectrum that may evolve over time, and includes classic kernicterus as well as other subtle forms of kernicterus, also known as bilirubin-induced neurological dysfunction or BIND) (Table 4).2,13,14 These manifestations include varying degrees of neurological deficits with different levels of cognitive impairment, learning disorders, attention disorders, abnormal movement disorders, or merely the hearing loss mentioned above.2,5,11,12
Clinical spectrum of bilirubin encephalopathy.
| “Classic” kernicterus |
| Acute bilirubin encephalopathy |
| Food rejection, lethargy, muscle tone alterations (hypotonia/hypertonia, retrocollis, opisthotonos, high-pitched cry, upward gaze, fever, seizures, coma, and death |
| BAEP abnormalities |
| MRI findings (T1): globi pallidi and subthalamic nuclei |
| Chronic bilirubin encephalopathy |
| Motor disorders: athetosis, dystonia, spasticity, or hypotonia |
| Auditory sensory disorder: hearing loss (total or partial) or auditory neuropathy |
| Oculomotor disorder: permanent vertical or lateral gaze |
| Dysplasia of the enamel of deciduous teeth: discolouration and loss of dental enamel |
| Subtle forms of kernicterus: bilirubin-induced neurological dysfunction (BIND) |
| Neurological impairments: cognitive, learning and motor disorders |
| Isolated hearing loss |
| Other potentially related complications: ADHD, autism, lack of coordination, Parkinson disease |
Kernicterus or bilirubin encephalopathy has been diagnosed by clinical means for many years. Improvements in both laboratory and neuroimaging diagnostic techniques and neurophysiological examinations, and the optimisation of such treatments as phototherapy and exchange transfusion, have led to drastic reductions in time to diagnosis. This means that treatment may be started earlier, which has resulted in a definite decrease in the incidence rate of this disease in recent years, although numbers may be on the rise once more. Among other factors, this is due to increased survival rates among pre-term neonates with an extremely low birth weight (very premature).2,5
At present, bilirubin encephalopathy in the full-term or nearly full-term neonate is defined not only by serum bilirubin levels, but also by the association of unconjugated bilirubin >20mg/dL and the presence of neurological anomalies (motor or sensory) or suggestive neuroimaging or BAEP findings.2,5
Typical pathological lesions are characterised not only by the yellowish colour in brain structures caused by regional bilirubin exudation (which may occur with no neuronal loss), but also by the association of selective neuronal affectation with residual gliosis of the subthalamic and globus pallidus areas.8,9,11 In the case of the patient who died, we discovered these typical signs using anatomical pathology methods. On the other hand, findings compatible with cortical hypoxia in our series are explained perfectly by the patient's clinical course of sepsis, coma and death secondary to multiple organ dysfunction. The patient remained on mechanical ventilation during the entire hospital stay. It is an interesting fact that few studies published to date correlate macroscopic findings with MRI images.8,9,11,12
With the development of MRI techniques, the brain lesions typical of this syndrome were identified and described in full-term neonates.2,12,15,17–20 Kernicterus lesions that can be detected by head MRI follow a chronological sequence. In the first phase (acute phase), we detect a signal increase in T1-weighted sequences in the globi pallidi and, at times, in the subthalamic nuclei. These alterations have been described in hyperbilirubinaemic pre-term or full-term neonates 5 days old.15 We believe that these findings reflect the immediate astroglial reaction to insult, oedema, or the presence of bilirubin.6,7,19,20 During a second (transitional) phase, the hyperintensity in the T1-weighted image decreases throughout the second and third weeks of life before becoming normal. In some patients, hyperintensity in T2-weighted images at the same location may be observed simultaneously, and may even extend to the cerebellum and brain stem.2 Increases in signal intensity in T2-weighted images have been described at 6 to 8 days after birth.7,12 Of our patients, 3 presented alterations in signal intensity in T2-weighted images (at 6 and 17 days of life); the remaining 3 did not show such changes at 7 days of life. Clinical course was unfavourable in all patients, with outcomes of death or major sequelae (Table 3). Neuroimaging findings evolved similarly in all cases observed. The hyperintensities in T1-weighted images practically disappeared, and then appeared in T2 images in the globi pallidi, and in 2 cases, the subthalamic nuclei (Figs. 2 and 4).
Lastly, during a third stage (the chronic phase), a hyperintense signal is identified in T2 which remains for the rest of the patient's life (Fig. 3), and has been found in children as old as 12 years.19 This signal is indicative of the dense fibrillar gliosis with a low cellular content appearing in the final phase, and which has been fully described since the 1970s.8,9
Neuroimaging findings do not exhibit 3 distinct phases in all patients. One published series describes 6 patients with initial anomalies in MRI images (hyperintensity in T1-weighted sequences only, in globi pallidi; subsequent MRI images were normal.18,19 Of these children, 4 (4/6) developed normally.15 We should state once again that not all patients with hyperbilirubinaemia, neurological symptoms in the neonatal period and positive neuroimaging results will develop the disease.5 This fact is extremely important in order to determine the prognosis in the neonatal period, or even to apply more or less aggressive treatment methods. However, such a result is quite rare, and when neonatal symptoms, biochemical parameters and neuroimaging and BAEP findings suggest bilirubin encephalopathy, the patient has a very high probability of developing the disease.
For this reason, we consider head MRI to be a useful tool for determining bilirubin toxicity in the central nervous system during the neonatal period. The presence of hyperbilirubinaemia associated with suggestive clinical symptoms and abnormal MRI findings (initially, hyperintensity in globi pallidi in T1 images, with subsequent hyperintensity in T2), together with abnormal BAEP activity, allows us to establish the diagnosis and a general prognosis and begin treatment.
Up until now, the only way of preventing such disorders was to measure serum bilirubin levels in clinically suspected cases and provide appropriate treatment where necessary. The use of international consensus protocols for treating hyperbilirubinaemia with phototherapy, exchange transfusion and additional measures (pre-exchange 5% albumin infusion), haem oxygenase inhibitors, enzyme inducers, inducing intestinal excretion of bilirubin, beta-glucuronidase inhibitors, intestinal chelation and others) will help reduce or minimise bilirubin toxicity to the central nervous system at these early ages.21–23
In conclusion, we must state that despite scientific advances, patients continue to suffer the sequelae of neonatal hyperbilirubinaemia, and that while our understanding of its physiopathological mechanisms has improved, it is not complete. In the past 10 years, we have observed an increase in the incidence of bilirubin encephalopathy. This may be attributed in part to the increase in complications typical of very premature and high-risk neonates, since these patients would not have survived the neonatal period a few years ago. Secondly, we are seeing an increased population of immigrants from developing countries where methods for monitoring bilirubin and treating high bilirubin levels are not in use. This also contributes to the increase in bilirubin encephalopathy rates. New neuroimaging techniques permit precise diagnosis of lesions in the basal ganglia, cerebellum and brain stem that are caused by hyperbilirubinaemia and provoke encephalopathy. Lastly, we should point out that many patients with sensorineural hearing loss of unknown aetiology may have suffered from the effects of hyperbilirubinaemia in the neonatal period. This may also be true in the case of minor neurological symptoms for which a probable cause cannot be determined.
Conflicts of interestThe authors have no conflicts of interest to declare.
articles

Neurología (English Edition) follows the Recommendations for the Conduct, Reporting, Editing and Publication of Scholarly Work in Medical Journals