



Benign epilepsy with central-temporal spikes (BECTS) is the most common epileptic syndrome in childhood. It is an age-dependent, genetically determined and benign condition. The aim of this study is to describe the clinical course and prognosis in 60 patients with BECTS diagnosed in our hospital.
Patients and methodsWe made a retrospective review of patients diagnosed with BECTS in a University Hospital (1995–2009). They were divided into 2 groups: (1) patients who met all BECTS classical criteria. (2) Patients who met all the criteria but one (less than 4 years; diurnal seizures; atypical EEG abnormalities).
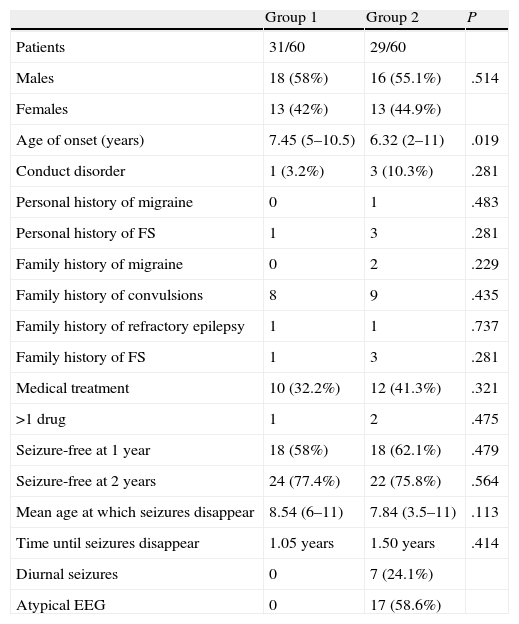
ResultsA total of 60 patients, 34 males and 26 females were included, with 31 patients in group 1 and 29 in group 2. The mean age at onset in group 1: 7.45 years, group 2: 6.55 years. Medical treatment was indicated in 32.2% of patients in group 1 and 41.3% in group 2. The outcome was favourable in the majority: 58% in group 1 and 62.1% in group 2 were free of seizures after 1 year. Average age in which it disappeared: 8.54 years in group 1 and 7.84 years in group 2. There were no statistically significant differences in any of these parameters.
ConclusionsUnlike that published by some authors, we have not identified any poor outcome factors in patients with BECTS in this study, meaning that an accurate diagnosis correlates with a good prognosis and excellent neurological outcome.
La epilepsia benigna con puntas centro-temporales (EBPCT) es el síndrome epiléptico más frecuente de la infancia, tiene carácter edad-dependiente, elevada predisposición genética y curso benigno. El objetivo de este trabajo es describir el curso clínico y el pronóstico de 60 pacientes diagnosticados de EBPCT en nuestro centro.
Pacientes y métodosRevisión retrospectiva de los pacientes diagnosticados de EBPCT en un hospital universitario (1995-2009). Se dividieron en 2 grupos: a) pacientes que cumplían todos los criterios clásicos de EBPCT, y b) cumplían los criterios excepto uno (menos de 4 años; crisis en vigilia; alteraciones EEG no típicas).
ResultadosSe seleccionó a 60 pacientes, 34 varones y 26 mujeres. Se incluyó a 31 pacientes en el grupo 1 y a 29 en el grupo 2. Edad media de inicio en el grupo 1: 7,45 años; grupo 2: 6,55 años. Se indicó tratamiento médico en 32,2% de pacientes del grupo 1, y en 41,3% del grupo 2. La evolución fue favorable en la mayoría: 58% en el grupo 1 y 62,1% en el 2 estaban libres de crisis tras un año. Edad media a la que desaparecieron: 8,54 años en el grupo 1 y 7,84 años en el grupo 2. No se encontraron diferencias estadísticamente significativas en ninguno de estos parámetros.
ConclusionesA diferencia de lo que algunos autores habían publicado, en este trabajo no se han identificado factores clínicos de mala evolución en pacientes con EBPCT, de modo que su diagnóstico se correlaciona con una evolución favorable y un excelente pronóstico neurológico.
Benign epilepsy with central-temporal spikes (BECTS) is considered the most common epilepsy syndrome in children, and accounts for 15%–24% of all paediatric epilepsy cases.1–4 Age of onset may vary greatly (2–13 years), with a mean age of 7 years. It is considered an age-dependent epileptic syndrome with a high genetic predisposition. Its course is benign.1–4
This type of epilepsy is diagnosed in previously neuropsychologically normal children with a phenotype typical of focal epilepsy primarily presenting during sleep. The typical ictal manifestation occurs with hemifacial clonic movements that may be preceded by somatosensory symptoms in the lips, tongue, and cheeks. It is sometimes accompanied by clonic movements of the ipsilateral upper and lower limbs2–4 and it occurs during sleep in 75% of cases, usually at the beginning or during the final stages (awakening).
Seizures normally happen in 20%–54% of cases and the characteristic EEG shows high-voltage spikes or spike-and-wave complexes in the centrotemporal region that may spread throughout the contralateral side. At the same time, background bioelectrical activity in the brain remains normal. Neuroimaging findings are normal.1–4
The aim of this study was to describe the clinical course and long-term prognosis in 60 patients with BECTS diagnosed in our hospital. We compared the characteristics of those patients who met all classical criteria with patients who met all the criteria but one.
Patients and methodsWe retrospectively reviewed the medical histories of patients diagnosed with BECTS in Hospital Virgen de la Arrixaca in Murcia between 1995 and 2009. We selected 60 patients for the retrospective analysis and eliminated the rest who did not meet our study's uniformity requirements due to lack of data and/or complementary tests. Evolution to atypical benign partial epilepsy was a criterion for exclusion. Analysed cases were divided into 2 groups. The first group included those patients who met all classical criteria for BECTS (nocturnal partial seizures with or without secondary generalisation; age of onset from 4 to 13 years and disappearance before 16 years; normal neurodevelopment; typical EEG with biphasic spikes in central-temporal areas, normal background activity and normal neuroimaging results). This group comprised 31 patients.
The second group included patients who met all the classical criteria of BECTS except for one of the following: age of onset before 4 years, predominantly diurnal seizures and EEG patterns that are not typical of BECTS. This group comprised 29 patients.
In all cases we recorded demographic data, accompanying disorders, medical and family history, neuroimaging findings, course of seizures and need for medication. The minimum follow-up period after diagnosis of epilepsy was 2 years.
Results were analysed with SPSS, using the Chi-square test for qualitative variables such as sex, presence of diagnosed conduct disorder, presence/absence of personal or family history of febrile seizures or migraine, family history of convulsions or refractory epilepsy, presence/absence of need for antiepileptic treatment, need for more than one drug, and seizure-free status after 1 and 2 years, respectively.
We used the Student t-test to compare age of onset and age of resolution of epilepsy between the 2 groups. We used the Mann–Whitney test to compare how much time elapsed before the disappearance of epileptic seizures. In both cases we used a P value of less than .05. Data from the statistical analysis are shown in Table 1.
Comparative analysis of the groups.
| Group 1 | Group 2 | P | |
| Patients | 31/60 | 29/60 | |
| Males | 18 (58%) | 16 (55.1%) | .514 |
| Females | 13 (42%) | 13 (44.9%) | |
| Age of onset (years) | 7.45 (5–10.5) | 6.32 (2–11) | .019 |
| Conduct disorder | 1 (3.2%) | 3 (10.3%) | .281 |
| Personal history of migraine | 0 | 1 | .483 |
| Personal history of FS | 1 | 3 | .281 |
| Family history of migraine | 0 | 2 | .229 |
| Family history of convulsions | 8 | 9 | .435 |
| Family history of refractory epilepsy | 1 | 1 | .737 |
| Family history of FS | 1 | 3 | .281 |
| Medical treatment | 10 (32.2%) | 12 (41.3%) | .321 |
| >1 drug | 1 | 2 | .475 |
| Seizure-free at 1 year | 18 (58%) | 18 (62.1%) | .479 |
| Seizure-free at 2 years | 24 (77.4%) | 22 (75.8%) | .564 |
| Mean age at which seizures disappear | 8.54 (6–11) | 7.84 (3.5–11) | .113 |
| Time until seizures disappear | 1.05 years | 1.50 years | .414 |
| Diurnal seizures | 0 | 7 (24.1%) | |
| Atypical EEG | 0 | 17 (58.6%) |
The 60 selected patients were divided into 2 groups, with similar numbers in each group: 31 patients in group 1 and 29 patients in group 2. Distribution by sex was also similar with 34 males and 26 females. Mean age of onset in the full sample was 7.01 years (range, 2.5–11.2 years). Age was lower in group 2 (6.55 years) than in group 1 (7.45 years), and the difference was statistically significant for a P value of .019. This finding was irrelevant and to be expected, since one of the inclusion criteria for group 2 was age of less than 4 years.
In 53 patients (88.3%) seizures normally occurred during sleep or near waking, and 7 of these patients (11.7%) also had seizures when awake (these 7 patients were included in group 2).
We observed that 4 patients had conduct disorder (including 2 cases of attention deficit hyperactivity disorder); 1 patient was in group 1 and 3 in group 2. This difference is not statistically significant. Cases of conduct disorder were mild and improved with cognitive behavioural therapy.
Four patients had a history of febrile seizures, 3 of them from group 2. A family history of migraine was reported in only 2 cases, both from group 2, and history of seizures in 8 patients from group 1 and 9 patients from group 2. Once again, these differences were not statistically significant.
Treatment was prescribed for 10 children in group 1 (32.2%) and 12 children in group 2 (41.3%). Multiple drugs were not needed in most cases (except in 1 patient in group 1 and 2 patients in group 2). The most widely used drug was carbamazepine, prescribed as the drug of choice in 8 patients, followed by valproic acid, the first choice in 6 patients. None of the examined patients suffered from cognitive impairment or experienced worsening of seizures because of medication. Two factors make the long-term follow-up of treated patients difficult: the retrospective nature of the study and the fact that patients were recruited in a regional neuropaediatric referral centre for children 11 and under. After that, children are attended in local hospitals, usually by general neurology departments.
In order to determine clinical course, we counted the number of patients who were seizure-free 1 year after diagnosis and the patients who were seizure-free after 2 years; the mean age at which seizures disappeared; and mean time elapsed between onset and resolution of epilepsy. Outcomes were favourable in most cases. In group 1, 58% of the patients were seizure-free after 1 year, and in group 2 that percentage was 62.1%. The mean age at which seizures disappeared was somewhat older in group 1 (8.54 years) than in group 2 (7.84 years), in spite of the fact that the mean time elapsed between onset and resolution was higher in group 2 (1.50 years) than in group 1 (1.05 years).
In these series we found no significant differences in either the number of patients who received treatment, or time elapsed before resolution of seizures. Each of the 60 patients had at least one EEG done while they were awake. In addition, 41 patients (68.3%) had this test repeated while they were sleeping.
Of the total, 43 patients (71.6%) presented a typical pattern, with high-voltage spikes or spike-and-wave complexes in the centrotemporal region that may or may not extend to the contralateral side. These 43 children include all 31 patients in group 1, plus 17 patients (58.6%) from the group with atypical BECTS. The most frequent abnormal findings were additional epileptic spikes in other areas (11 patients), normal pattern (3 patients) and other EEG findings (another 3 patients). The patients with normal EEG findings had infrequent seizures (exclusively nocturnal seizures, between 2 and 4 throughout the course of the disease). They had no conduct disorders, no additional events, and none experienced seizures after the age of 8.
A neuroimaging study performed in 42 patients (70%) delivered normal results, except for 1 patient who presented an Arnold–Chiari malformation (type 1) as an incidental finding. Obviously, this was unrelated to the cause of the epileptic seizures. Demographic data, the clinical course, and comparisons between groups are shown in Table 1.
DiscussionAtypical BECTS findings may be classified according to seizure type (exclusively diurnal seizures, postictal paralysis, prolonged seizures) or EEG patterns (atypical spike morphology, unusual location, spike-and-wave discharges or abnormal background activity), although early age of onset (less than 4 years) seems to be the chief factor determining atypical course of the syndrome.5–8 Some authors have linked this form of epilepsy to a higher probability of learning difficulties and behavioural disorders.9
The clinical aspects and the EEG characteristics of the children included in this study coincide with the previous literature, with an age of onset between 2.5 and 11.2 years.
Neuropsychological development in children with BECTS is normal, with fewer behavioural disorders than are seen in other forms of paediatric epilepsy. Nevertheless, in recent years, studies have been published describing a higher probability of language impairment and/or reading and writing disorders, learning disabilities and/or attention deficit disorder in these patients.9–12
In fact, some authors4,6,13 support the idea that BECTS and its different atypical variants (including epilepsy with continuous spikes and waves during slow-wave sleep and Landau–Kleffner syndrome) are part of a spectrum with the same genetic basis, but differ in the duration, location and diffusion of epileptic activity.
Some of the atypical forms may give rise to atypical evolutions which are triggered by some antiepileptic drugs.4,6,13
Echoing earlier studies, our study recorded a high incidence (50%) of patients who did not meet all classical criteria for BECTS. This finding had to do with criteria used to define the clinical variants, which differ from author to author. For example, Verrotti et al.9 include patients with other types of seizures in the “atypical” group; Tavares et al.8 include patients with other types of seizures, patients with exclusively diurnal seizures and atypical EEG patterns; Datta and Sinclair5 and Callenbach et al.7 include patients younger than 4 years with learning disabilities, abnormal neurological findings, and other types of seizures and/or anomalies in the EEG. We used the last set of inclusion criteria.
Some of the cited studies5,7–9 attempted to identify factors predicting prognosis, but no factors have been clearly linked to worse neuropsychological outcome or longer duration of the epileptic syndrome. In the Datta and Sinclair5 series, the atypical group experienced seizures that were more difficult to control, but prognosis was the same. In the Verrotti et al.9 series, the atypical group experienced more learning and conduct disorders, but these results have not been confirmed by other authors.7,8 Our study found a higher proportion of patients with conduct disorders in group 2, but the difference was not statistically significant. It will be necessary to perform more in-depth prospective studies in order to corroborate or disprove this result. Given that the outlook for most of these patients is excellent, it is difficult to identify patients whose outcome will be less favourable. Moreover, the results of our study do not allow us to establish a poorer theoretical prognosis for one of the BECTS groups, since course of disease was very similar in both groups.
Conflicts of interestThe authors have no conflicts of interest to declare.
We would like to thank Dr Juan Ignacio Ortuño Sempere, paediatrician at Hospital Los Arcos del Mar Menor, for his assistance with the statistical study.
Please cite this article as: Ibáñez Micó S, et al. Variantes clínicas de epilepsia rolándica y su implicación en el pronóstico. Neurología. 2012;27:212–5.
