





Charcot-Marie-Tooth disease (CMT) is the most frequent form of inherited neuropathy. In accordance with the inheritance pattern and degree of slowing of motor conduction velocity (MCV) of the median nerve, CMT encompasses five main forms: CMT1 (autosomal dominant [AD] or X-linked transmission and MCV<38m/s); CMT2 (AD or X-linked transmission and MCV>38m/s); CMT4 (autosomal recessive [AR] and severe slowing of MCV); AR-CMT2 (AR transmission and MCV>38m/s); and DI-CMT (intermediate form with AD transmission and MCV between 30 and 40m/s). In spite of its stereotyped semiological repertoire (basically, symptoms and signs of sensory-motor polyneuropathy and pes cavus), CMT seems to be one of the most complex hereditary neurodegenerative syndromes, 31 causative genes having been cloned.
DevelopmentThis paper is aimed at performing a nosological review of the disease, emphasising the guidelines for its molecular diagnosis. Genetic epidemiological studies and genotypes reported in Spanish patients are revised.
ConclusionsIn the great majority of CMT cases, mutations involve a reduced number of genes, namely: for CMT1, PMP22, GJB1 and MPZ; for CMT2, MFN2 and GJB1; for CMT4, GDAP1, and NDRG1, HK1 and SH3TC2 (gypsies); for AR-CMT2, GDAP1; and for DI-CMT, GJB1 and MPZ. Given their low prevalence, mutations in other pathogenic genes should be investigated after discarding the previous ones. There is no place for the indiscriminate use of diagnostic CMT genetic panels.
La enfermedad de Charcot-Marie-Tooth (CMT) es la neuropatía hereditaria más frecuente. Clásicamente dividida según su patrón de herencia y de alteración de la velocidad de conducción motora (VCM) del nervio mediano, CMT incluye cinco grandes categorías: CMT1 (herencia autosómica dominante [AD] o ligada al sexo, y VCM<38m/s); CMT2 (herencia AD o ligada al sexo y VCM>38m/s); CMT4 (herencia autosómica recesiva [AR] y VCM muy lentificada); AR-CMT2 (forma recesiva con VCM>38m/s), y DI-CMT (forma intermedia con herencia AD y VCM entre 30 y 40m/s). Pese a su estereotipado cuadro clínico (básicamente, semiología polineuropática sensitivo-motora y pie cavo), CMT ha resultado ser uno de los síndromes neurodegenerativos genéticamente más complejos, con 31 genes patogénicos clonados.
DesarrolloEl objetivo de esta guía es efectuar una revisión nosológica de la enfermedad de CMT, con énfasis en las directrices para llevar a cabo el diagnóstico molecular. A tal fin, revisamos los estudios de epidemiología y genética, y los genotipos descritos en España.
ConclusionesEn la inmensa mayoría de los pacientes con CMT, las mutaciones recaen en un reducido número de genes: para CMT1, PMP22, GJB1 y MPZ; para CMT2, MFN2 y GJB1; para CMT4, GDAP1, y NDRG1, HK1 y SH3TC2 (sujetos de etnia gitana); para AR-CMT2, GDAP1, y para DI-CMT, GJB1 y MPZ. Por su baja prevalencia, las mutaciones en otros genes sólo deberían investigarse cuando las anteriores han sido descartadas. Se desaconseja el uso indiscriminado de paneles de múltiples genes para el diagnóstico molecular de la enfermedad.
Charcot-Marie-Tooth (CMT) disease is the most common hereditary neuropathy, with a prevalence in Spain of 28.2 cases per 100000 inhabitants.1 In general, this syndrome has an infantile or juvenile onset, with motor and sensory polyneuropathic semiology and pes cavus.2–5 CMT can be transmitted through autosomal dominant (AD), autosomal recessive (AR) or X-linked inheritance. According to median nerve motor conduction velocity values (MCV), there are demyelinating forms (MCV<38m/s), axonal forms (MCV>38m/s) and intermediate forms (MCV=30–40m/s).2–4 In good correlation with neurophysiological descriptions, histological studies of the peripheral nervous system (PNS) have demonstrated a dual pattern, either axonal or demyelinating. Based on these clinical, neurophysiological and pathological data, Dyck proposed a simple, unanimously accepted classification in the 1970s. This classification included the following types: a) type I (CMT1, demyelinating or hypertrophic) with AD or AR inheritance; b) type II (CMT2, neuronal or axonal) with AD or AR inheritance; c) type III (CMT3, usually with AR inheritance) reserved for Dejerine–Sottas disease or patients with severe forms of hypomyelinating CMT; d) X-linked forms, and e) complex forms (e.g. with optic atrophy, deafness or pigment degeneration of the retina).5 Although in the literature this disease has also been designated as hereditary motor and sensory neuropathy (HMSN), at present the acronym CMT is more commonly used. It is worth noting that the only clinical sign which differentiates CMT1 from CMT2 is the presence of visible or palpable thickening of the nerve trunks in CMT1.
The prodigious advances in molecular genetics over the past 2 decades have meant that the nosology of CMT has been constantly changing, to the extent that this disease includes over 30 cloned pathogenic genes (see below). This guide is an initiative of Programme 3 (Neuromuscular Diseases) at CIBERNED, which aims to conduct a brief nosological review of the disease, as well as an analysis of disease diagnosis with emphasis on the selection of molecular tests.
Charcot-Marie-Tooth disease in the molecular eraBefore starting this section, we must remember that CMT has a very close nosological relationship with 2 other forms of hereditary neuropathy: distal hereditary motor neuronopathy (dHMN) and hereditary sensory and autonomic neuropathy (HSAN). Among these 3 syndromes there is not only a phenotypic overlap, but also the phenomenon of allelic heterogeneity (identical phenotype caused by different mutations in the same gene and chromosomal locus) and locus heterogeneity (mutations in genes in different chromosomal loci giving rise to the same phenotype). For the sake of brevity, we will only deal with CMT and only refer to HSAN and dHMN when appropriate.
A total of 37 loci with 31 cloned genes have been identified using genetic linkage analysis.3,4,6–8 An additional 12 loci with 9 cloned genes have been described in HMN/HSAN (see reference 4 for review). It is estimated that, overall, the molecular basis of 33% of CMT cases still remains to be discovered.9 This challenge may be aided by new, whole-genome sequencing techniques.10,11 These genes and their respective proteins constitute a microarray of molecules which are necessary for the normal functioning of the PNS.6 It is somewhat ironic that, despite the apparent simplicity of its semiotic repertoire, CMT has emerged as one of the most genetically complex neurological syndromes.
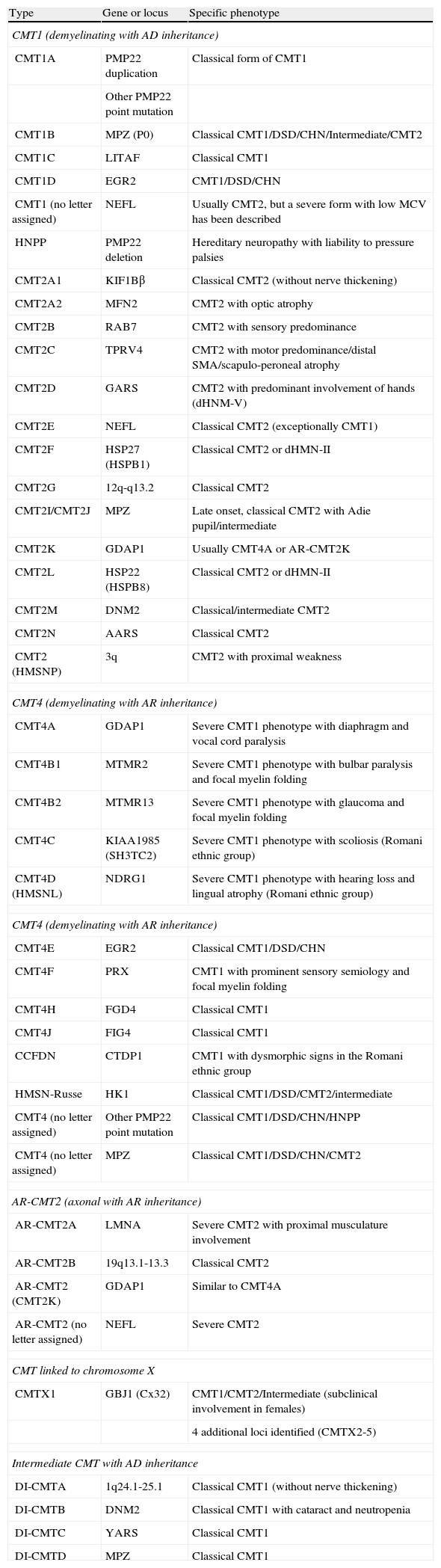
Table 1, adapted from references 4, 7 and 8, shows an updated clinical-genetic classification of CMT, which is tentative since there is no unanimous view on the use of its types and subtypes. There is universal agreement in accepting CMT1 as a header for demyelinating phenotypes with AD inheritance. Meanwhile, some authors include axonal forms with AD or AR inheritance in CMT2, while others include only AD forms, thus creating the acronym AR-CMT2 for axonal forms with AR transmission. We have followed this approach. The acronym CMT3, applied in the Dyck classification5 to syndromes similar to that described by Dejerine and Sottas,12 disappears and is replaced by CMT4, which encompasses all demyelinating syndromes with AR inheritance. In short, the acronym DI-CMT is introduced for intermediate forms with AD transmission.
Clinical-genetic classification of CMT.
| Type | Gene or locus | Specific phenotype |
| CMT1 (demyelinating with AD inheritance) | ||
| CMT1A | PMP22 duplication | Classical form of CMT1 |
| Other PMP22 point mutation | ||
| CMT1B | MPZ (P0) | Classical CMT1/DSD/CHN/Intermediate/CMT2 |
| CMT1C | LITAF | Classical CMT1 |
| CMT1D | EGR2 | CMT1/DSD/CHN |
| CMT1 (no letter assigned) | NEFL | Usually CMT2, but a severe form with low MCV has been described |
| HNPP | PMP22 deletion | Hereditary neuropathy with liability to pressure palsies |
| CMT2A1 | KIF1Bβ | Classical CMT2 (without nerve thickening) |
| CMT2A2 | MFN2 | CMT2 with optic atrophy |
| CMT2B | RAB7 | CMT2 with sensory predominance |
| CMT2C | TPRV4 | CMT2 with motor predominance/distal SMA/scapulo-peroneal atrophy |
| CMT2D | GARS | CMT2 with predominant involvement of hands (dHNM-V) |
| CMT2E | NEFL | Classical CMT2 (exceptionally CMT1) |
| CMT2F | HSP27 (HSPB1) | Classical CMT2 or dHMN-II |
| CMT2G | 12q-q13.2 | Classical CMT2 |
| CMT2I/CMT2J | MPZ | Late onset, classical CMT2 with Adie pupil/intermediate |
| CMT2K | GDAP1 | Usually CMT4A or AR-CMT2K |
| CMT2L | HSP22 (HSPB8) | Classical CMT2 or dHMN-II |
| CMT2M | DNM2 | Classical/intermediate CMT2 |
| CMT2N | AARS | Classical CMT2 |
| CMT2 (HMSNP) | 3q | CMT2 with proximal weakness |
| CMT4 (demyelinating with AR inheritance) | ||
| CMT4A | GDAP1 | Severe CMT1 phenotype with diaphragm and vocal cord paralysis |
| CMT4B1 | MTMR2 | Severe CMT1 phenotype with bulbar paralysis and focal myelin folding |
| CMT4B2 | MTMR13 | Severe CMT1 phenotype with glaucoma and focal myelin folding |
| CMT4C | KIAA1985 (SH3TC2) | Severe CMT1 phenotype with scoliosis (Romani ethnic group) |
| CMT4D (HMSNL) | NDRG1 | Severe CMT1 phenotype with hearing loss and lingual atrophy (Romani ethnic group) |
| CMT4 (demyelinating with AR inheritance) | ||
| CMT4E | EGR2 | Classical CMT1/DSD/CHN |
| CMT4F | PRX | CMT1 with prominent sensory semiology and focal myelin folding |
| CMT4H | FGD4 | Classical CMT1 |
| CMT4J | FIG4 | Classical CMT1 |
| CCFDN | CTDP1 | CMT1 with dysmorphic signs in the Romani ethnic group |
| HMSN-Russe | HK1 | Classical CMT1/DSD/CMT2/intermediate |
| CMT4 (no letter assigned) | Other PMP22 point mutation | Classical CMT1/DSD/CHN/HNPP |
| CMT4 (no letter assigned) | MPZ | Classical CMT1/DSD/CHN/CMT2 |
| AR-CMT2 (axonal with AR inheritance) | ||
| AR-CMT2A | LMNA | Severe CMT2 with proximal musculature involvement |
| AR-CMT2B | 19q13.1-13.3 | Classical CMT2 |
| AR-CMT2 (CMT2K) | GDAP1 | Similar to CMT4A |
| AR-CMT2 (no letter assigned) | NEFL | Severe CMT2 |
| CMT linked to chromosome X | ||
| CMTX1 | GBJ1 (Cx32) | CMT1/CMT2/Intermediate (subclinical involvement in females) |
| 4 additional loci identified (CMTX2-5) | ||
| Intermediate CMT with AD inheritance | ||
| DI-CMTA | 1q24.1-25.1 | Classical CMT1 (without nerve thickening) |
| DI-CMTB | DNM2 | Classical CMT1 with cataract and neutropenia |
| DI-CMTC | YARS | Classical CMT1 |
| DI-CMTD | MPZ | Classical CMT1 |
AARS: alanyl tRNA synthetase; CHN: congenital hypomyelinating neuropathy; CMT: Charcot-Marie-Tooth disease; CTDP1: CTD phosphatase subunit 1; DNM2: dynamin 2; DSD: Dejerine–Sottas disease; EGR2: early growth response 2; FDG4: RhoGEF; FIG4: Ptdlns(3,5)P25-phosphatase; GARS: glycyl tRNA synthetase; GBJ1: gap junction protein beta 1; GDAP1: ganglioside induced differentiation associated protein 1; HK1: hexoskinase 1; HMSNL: hereditary motor and sensory neuropathy Lom; HNPP: hereditary susceptibility pressure palsy; HSP22: heat shock 22kDa protein; HSP27: heat shock 27kDa protein; KIF1Bβ: kinesin family member 1-Bβ; LITAF: lipopolysaccharide induced tumour necrosis factor; LMNA: lamin A/C; MFN2: mitofusin 2; MTMR2: myotubularin related protein 2; MTMR13: myotubularin related protein 13; NDRG1: N-myc downstream regulated gene; NEFL: neurofilament light polypeptide 68kDa; PMP22: peripheral myelin protein 22; P0: myelin protein zero; PRX: periaxin; RAB7: RAB7, member RAS encogen family; SH3TC2: SH3 domain and tetratricopeptide repeats; SMA: spinal muscular atrophy; TRPV4: transient receptor potential vallinoid 4; YARS: tyrosyl tRNA synthetase.
See http://neuromuscular.wustl.edu/time/hmsn.html.
Fig. 1 illustrates the location of the mutated proteins, which was predictable for those known components of the PNS, such as proteins PMP22 and MPZ (P0) of compact myelin. However, in other cases the discovery of the mutated pathogenic protein proved unexpected. One example is illustrated by the case of GDAP1, whose function in the PNS was unknown until the identification of CMT4A took place.13,14
, HSN2 (hereditary sensory neuropathy type 2), NTRK1 (neurotrophic tyrosine kinase receptor type 1), IKBKAP (inhibitor of kappa light poypeptide gene enhancer in B-cells) and NGF1 (nerve growth factor beta polypeptide) are involved in the aetiopathogenesis of hereditary sensory and autonomic neuropathies, which are not reviewed here (see text).")
Image of a myelinated nerve fibre. The mutated proteins which cause CMT, HMN or HSAN identified up to 2006 are shown in black, while those described subsequently are shown in red. The meaning of acronyms is given at the bottom of Table 1.
Note that mutations of SPTLC1 (serine palmitoyltransferase long chain base subunit 1), HSN2 (hereditary sensory neuropathy type 2), NTRK1 (neurotrophic tyrosine kinase receptor type 1), IKBKAP (inhibitor of kappa light poypeptide gene enhancer in B-cells) and NGF1 (nerve growth factor beta polypeptide) are involved in the aetiopathogenesis of hereditary sensory and autonomic neuropathies, which are not reviewed here (see text).
From an educational point of view and according to Niemann et al,6 the aetiopathogenic mechanisms of the mutated proteins are summarised as follows: a) alteration of the development and maintenance of myelin; b) alteration of the biosynthesis and degradation of proteins; c) alteration of the endocytosis and dynamics of membranes, including the mitochondrial membrane; d) alteration of the axonal cytoskeleton; e) seipinopathies,15 and f) channelopathies by mutation of TRPV4.16 We will briefly review these 6 sections. In CMT1/CMT4 forms by mutation of certain myelin components, it is assumed that the Schwann cell defect causes demyelination/dysmyelination with secondary axonopathy, which is ultimately responsible for the clinical semiology.17–20 The most common syndrome in this section is CMT1A, which accounts for 55% of all CMT cases and 66.8% of CMT1 cases,9 and which is usually caused by an allelic trisomy of 17p11.2 of 1.5Mb, containing the gene PMP22.21,22 This trisomy causes an excessive gene dosage, leading to overproduction of PMP22 and its accumulation in Schwann cells, inducing endoplasmic reticulum stress, resulting in programmed cell death. Deletion works by reducing the expression of PMP22, producing unstable myelin which is manifested as hereditary neuropathy with liability to pressure palsies (HNPP) (Table 1). In a small percentage of cases, duplication/deletion can occur as a de novo phenomenon. Other point mutations (e.g. single-base substitutions) of the PMP22 gene are rare and cause severe phenotypes, either AD (probably through a function gain mechanism) or AR (loss of function due to failure in the synthesis of PMP22).23 Protein MPZ is quantitatively the most abundant within compact myelin and an essential element for its compaction. In 10% of cases, CMT is caused by point mutations of MPZ resulting in either a demyelinating, early-onset AD phenotype (CMT1B) or, exceptionally, an AR phenotype, or else in an axonal, late-onset phenotype (CMT2I and CMT2J).24,25 Thus, the molecular pathology of PMP22/MPZ unveiled that their mutations can be inherited through both AD and AR transmission, and that, in the case of MPZ, its mutations cause both a demyelinating and axonal phenotype. This only highlights the fact that communication between Schwann cells and accompanying axons within the PNS is continuous.6 Such phenomena are applicable to mutations causing CMT in other genes (Table 1). GBJ1 (Cx32) is a gap-type protein of paranodal myelin, whose gene is located on chromosome X. Point mutations in the GBJ1 gene are the second most common cause of CMT and cause dysfunction in the radial transit of small molecules between Schwann cells and axons.6,26,27 Probably by a haploinsufficiency mechanism, such mutations cause a more severe phenotype in males than in females, which neurophysiologically can be demyelinating, axonal or intermediate. Other, rarer causes of CMT1/CMT4 include mutations in the EGR2 gene (which encodes a transcription factor involved in regulating myelin genes) and the PRX gene (which encodes a cytoskeletal anchoring protein of Schwann cells).
The correct composition and maintenance of the membranous compartments of Schwann cells and PNS neurons depend on a perfect balance between the synthesis of structural and signalling components and their degradation processes.6 Among the mutated proteins involved in endocytosis processes are the following (Table 1): a) phosphatases (MTMR2, MTMR13 and FIG4), which cause severe AR phenotypes (CMT4B1, CMT4B2 and CMT4J) with focal myelin folds (CMT4B1 and CMT4B2); b) GTPases: DNM2 with an AD phenotype that can be either intermediate (DI-CMTB) or axonal,28,29 RAB7 which causes CMT2B (a phenotype similar to that of HSNA1), and FRABIN which is associated with CMT4H. Mutations of NDRG1, a regulatory gene whose function is poorly known, cause a severe syndrome (CMT4D) in subjects from the Romani ethnic group. With regard to the components involved in the synthesis, classification and degradation of proteins, the mutations affect the following components: a) LITAF/SIMPLE, an ubiquitin ligase, which causes CMT1C, and b) GARS, YARS and AARS, proteins involved in the loading of tRNA with glycine, tyrosine and alanine, which originate CMT2D/dHMN-V, DI-CMTC and CMT2N, respectively (Table 1).
PNS neurons, both sensory and motor, must move proteins, vesicles and organelles through the long axonal stretches ranging from the soma to their terminals, and so require a complex and efficient transport system. Thus, the growing number of axonal CMT forms caused by mutations in proteins related to the cytoskeleton and to protein, vesicle and organelle transport (Table 1) should not be surprising.30 Mutations in the neurofilament light chain (NEFL) cause CMT2E and, exceptionally, CMT1F. Heat shock proteins (HSP) are ubiquitous macromolecules which, in the PNS, control the assembly of neurofilaments. Mutations of the HSP27 gene cause CMT2F/dHMN-II, while mutations in the HSP22 gene are associated with CMT2L/dHMN-II. Recently, a CMT line associated with the mutation HSP27 R127W, comprised by 10 patients who had been examined clinically and neurophysiologically, presented some cases with CMT2 phenotype and others with HMN phenotype. This highlights the fact that both syndromes may be a single nosological entity.31 Kinesins are a family of motor proteins which mediate in anterograde axonal transport on microtubules, while dyneins mediate retrograde transport. Mutations of the KIF1Bβ gene are associated with CMT2A1 and mutations of the RAB7 gene, encoding a GTPase which regulates dynein function, cause CMT2B. Mitochondrial morphology is determined by a balance between fusion and fission processes of the organelle.32 MNF2 is a GTPase of the mitochondrial outer wall, where it acts as a regulator of mitochondrial fusion. Point mutations in the MFN2 gene cause CMT2A2, which is currently the most common form of CMT2 (20%), with one fifth of cases appearing as de novo mutations.33–35 Mimicking HMSN-VI, optic atrophy may appear in CMT2A2, especially in severe forms with early onset. GDAP1 is the counterpart of MFN2, participating in mitochondrial fission processes. Homozygous mutations of GDAP1 cause either CMT4A or AR-CMT2.13,14,36,37 Exceptionally, certain mutations in this gene cause disease in a heterozygous state (CMT2K).38,39 The LMNA gene encodes a nuclear membrane protein whose mutation is associated with AR-CMT2A; it is interesting to note that mutations in this gene can cause Emery-Dreifuss myopathy. The KIAA1985/SH3TC2 gene encodes an adapter protein and its mutations cause a severe phenotype (CMT4C) (Table 1).10
BSCL2 is an acronym derived from Berardinelli-Seip congenital lipodystrophy 2, a syndrome originally described in strains with lipoatrophy, insulin resistance, hypertriglyceridemia, mental retardation and AD inheritance. BSCL2 or seipin is a glycosylated protein of the endoplasmic reticulum, whose mutations activate the UPR pathway (unfolded protein response), inducing endoplasmic reticulum stress and programmed cell death.15 Seipinopathies are considered a new model of disease by alteration of protein conformation. Point mutations of BSCL2 cause a continuous spectrum of neurodegenerative syndromes with AD transmission, including dHMN-V, Silver syndrome (spastic paraparesis and hand amyotrophy), CMT2 and hereditary spastic paraparesis. In a considerable percentage of cases, the mutation displays incomplete penetrance.40
TRPV4 is a nonselective cation channel involved in the detection of physical and chemical stimuli which takes part in multiple physiological functions.41 Heterozygous mutations of the TRPV4 gene have been associated with bone dysplasias. Genetic linkage analysis has shown that CMT2C, the scapulo-peroneal form of spinal muscular atrophy (SMA) and the congenital, distal form of SMA could be allelic syndromes (12q21-q24). Indeed, recent studies have shown that these syndromes are associated with different, heterozygous, point mutations in the ankyrin domain of TRPV4, sometimes with incomplete penetrance.16,42–45 The mechanism by which these mutations cause degeneration of the PNS is unknown. In any case, the disease is a prototypical example of variable inter- and intrafamilial expression.16,45
Clinical diagnosis of Charcot-Marie-Tooth diseaseThe first step is to establish whether the patient suffers a hereditary neuropathy. The answer may be obvious when the family survey shows a lineage with affected ancestors, suggesting AD or gender-linked inheritance (when there is no male-to-male transmission). The occurrence of disease among siblings and parental consanguinity suggest an AR inheritance. However, sometimes the family survey is negative, in which case there are a number of factors which may point towards genetic neuropathies, namely: a) onset during childhood; b) prolonged and slowly progressive clinical course; c) presence of pes cavus, and d) unlike in acquired neuropathies, absence of positive sensory symptoms (paraesthesias or dysaesthesias) despite a clear semiology of sensory deficit.8 Since affected individuals often present subtle symptoms or are even asymptomatic, it is also important to test the maximum possible number of subjects at risk within the lineage (secondary cases). This will enable detection of minimal signs of disease (e.g. arreflexia or pes cavus) in subclinical cases and, thus, help define the hereditary pattern more accurately.46 Examination of the musculature of the lower limbs using MRI has helped to detect early signs of fat atrophy in the intrinsic muscles of the feet.39,47,48
The next step should be a neurophysiological examination, including determination of the MCV and SCV (sensory conduction velocity) in at least 3 nerves. Following the guidelines outlined by the Charcot-Marie-Tooth Neuropathy Score (CMTNS) is advisable.49 The amplitude of compound motor action potentials (CMAP) should be taken into consideration when interpreting the degree of slowness in the MCV. A sharp drop in distal CMAP amplitude implies a loss of thick fibres depending on the distance, and this may involve a proportional reduction in the MCV. Examination of proximal nerve segments is recommended, in order to discern between MCV drops due to axonopathy or to myelinopathy. Conduction at these points will be similarly slowed down in cases of demyelinating CMT and less slowed down or even preserved in cases of axonal CMT.17–19,50 The slowing of MCV/SCV in CMT1/CMT4 is diffuse and uniform, and both CMAP morphology and the terminal latency index are usually preserved, in contrast to cases of acquired inflammatory neuropathy.19 In intermediate forms, the MCV is between 30 and 40m/s (or 25 and 45, according to some authors) in nerve trunks with both reduced CMAP (usually distal segments) and preserved CMAP (usually proximal segments). In other words, the causal gene mutation acts by originating a dysfunction of both the axon and the Schwann cell.51
Currently, nerve biopsy is reserved for cases where there are problems in the differential diagnosis with other hereditary neuropathies (e.g. amyloidosis) or with acquired neuropathies.
Molecular diagnosis of Charcot-Marie-Tooth diseaseOut of over 30 pathogenic genes identified so far (Table 1), only a small and variable proportion of them are available in Spain for study in molecular genetics laboratories, either from public hospitals or private entities. The cost of molecular diagnosis in our environment is covered by the Ministry of Health through the Cohesion Fund Information System (SIFCO). Given the enormous and growing number of pathogenic genes involved in CMT and the considerable cost of molecular studies, it is clear that genetic testing must be specifically targeted. A recent communication from the American Academy of Neurology recommends that the molecular diagnosis of CMT be carried out based on the clinical phenotype, inheritance pattern and neurophysiological findings, starting with the analysis of PMP22 duplication/deletion in the case of demyelinating phenotypes with AD inheritance or GJB1 or MFN2 mutations in the case of axonal phenotypes with vertical inheritance.52 Given the enormous molecular complexity of CMT, genetic epidemiology studies are scarce.9,53,54 We will refer to the work of Saporta et al,9 as it is the most recent, extensive, and detailed.
These authors reviewed cases referred to their monographic CMT consultation between 1997 and 2007.9 Their results included 1024 patients, of whom 787 were diagnosed with CMT. A total of 527 patients (67%) were categorised genetically and the remaining 260 were not categorised as CMT. Among the 527 cases defined genetically, the most common subtypes were: CMT1A (55%; PMP22 duplication), CMT1X (15.2%; GJB1 mutation), HNPP (9.1%; PMP22 deletion), CMT1B (8.5%; MPZ mutation) and CMT2A (4%; MFN2 mutation). Other mutations were detected in 23 patients (4.4% of total CMT cases defined genetically). These affected CMT1 or CMT2 patients and were distributed as follows: CMT1C (LITAF), 5 cases; CMT1D (EGR2), 1 case; CMT1E (PMP22 point mutation), 5 cases; CMT2D (GARS), 3 cases; CMT2E (NEFL), 4 cases, and CMT2K (GDAP1), 5 cases. In this study, only 1.8% of patients with CMT1 were not genetically categorised, while the percentage of such cases for CMT2 was 65%. Only 7 patients (1.4%) presented demyelinating, autosomal recessive forms (CMT4), with the following distribution: CMT4A (GDAP1), 1 case; CMT4C (SH3TC2), 3 cases; CMT4F (PRX), 1 case, and CMT4J (FIG4), 2 cases.
The work of Saporta et al also helps establish the percentages of success for each mutation studied. These range between 80% for PMP22 and 13% for MFN2.9 Based on certain phenotypic traits and the neurophysiological study, the authors provide several algorithms for the selection of molecular analysis, with the intention of enhancing diagnostic confidence. On the basis of these algorithms and our own experience, we designed a comprehensive diagnostic algorithm for all forms of CMT (Fig. 2). Below, we discuss some peculiarities which facilitate molecular diagnosis in CMT with vertical transmission:
- –
Given that 89% of patients with classical phenotype, without delay in the onset of walking (<15months), AD inheritance and MCV in arms between 15 and 35m/s present CMT1A, the first and only genetic test to be performed in case of a CMT1 syndrome should be PMP22 duplication.9 If negative, then GBJ1 and MPZ should be analysed; mutations in other genes which cause CMT1 are exceptional (see above and Fig. 2).
- –
In patients with similar characteristics to the previous paragraph but with very slow MCV (<15m/s), molecular analysis may start with PMP22 or MPZ, although delay in the onset of walking occurs more often in CMT1B that in CMT1A.9,24
- –
Mutation of GJB1 (CMT1X) may be associated with MCV within the limits established for demyelinating CMT, axonal CMT or intermediate CMT. Due to their high prevalence, connectopathies should be considered in all CMT syndromes with apparent vertical inheritance but no evidence of male–male transmission (Fig. 2).9,27
- –
The molecular study of patients with axonal CMT and AD inheritance should start with MFN2, MPZ and GJB1.
- –
The association with optic atrophy is characteristic of early onset CMT2A2 (Table 1).33 A late onset phenotype with Adie pupil should point towards CMT2J.25 Although the probability of detecting mutations in the remaining 11 genes is low (Fig. 2), there have been reports of Spanish CMT2 strains with mutations in DNM2, GDAP1 and TRPV4.16,29,39,45 Moreover, in our laboratories we have detected occasional, unpublished cases of CMT2 associated with mutations in NEFL, HSP27/HSPB1 and HSP22/HSPB22.
. AD: autosomal dominant; AR: autosomal recessive; ♂→♂: male–male transmission; ♂→|♂: no evidence of male–male transmission.")
Diagnostic algorithm for patients with CMT. Genetic mutations described both in the work of Saporta et al9 and in Spanish patients are shown in bold (see text for details, especially in relation to the priorities of molecular study).
AD: autosomal dominant; AR: autosomal recessive; ♂→♂: male–male transmission; ♂→|♂: no evidence of male–male transmission.
CMT with intermediate MCV and AD transmission (DI-CMT in Table 1) includes 4 different forms. The first has no definite molecular basis and the remaining 3 are linked to mutations in DNM2, YARS and MPZ. In our environment, there have been reports of intermediate phenotypes caused by mutations in MPZ.55 As previously noted, mutations in GJB1 should be included here. It is worth noting that in the series described by Saporta et al,9 the 2 causes of DI-CMT were mutations in GJB1 and MPZ.
CMT forms with AR transmission represent about 4% of the European CMT population, although in countries with high consanguinity, such as those in the Mediterranean and Middle East regions, the percentages of AR-CMT can reach between 30% and 50% of all CMT cases.56 Information on the genetic epidemiology of these regions is essential to prioritise the molecular study of the 14 genes underlying AR forms, whether demyelinating (CMT4) or axonal (AR-CMT2). In general (Table 1), recessive forms have a childhood onset and are more severe than dominant phenotypes, whilst CMT4 forms present a marked decrease in MCV (usually <15m/s).4 The eponyms Dejerine–Sottas disease and congenital hypomyelinating neuropathy probably apply to these recessive demyelinating forms.57,58 The founding mutations of NDRG1, SH3TC2 and HK1, all 3 identified in Spain, must be considered in patients from the Romani ethnic group.59–62 In our environment, GDAP1 mutation is the most frequent cause of recessive CMT, whether CMT4A or AR-CMT2 (CMT2K).36,37 This is a severe, childhood-onset phenotype, with proximal muscle involvement and diaphragmatic and vocal cord paralysis. It is interesting that GDAP1 heterozygous mutations can also be associated with a less severe axonal phenotype with AD transmission.39 The same phenomenon, co-participation of AR and AD inheritance patterns, has been exceptionally described for mutations of NEFL, HSP22 and MFN2.63–66Table 1 and Fig. 2 show other mutations associated with CMT with an AR transmission.
ConclusionThe diagnosis of CMT should be based on an adequate clinical, family and neurophysiological study. This will enable the CMT phenotype (CMT1, CMT2, CMT4, AR-CMT2 or DI-CMT) to be established with some certainty. With this clinical background, and for the vast majority of patients, genetic analysis can be directed towards the search for mutations within a reduced group of genes, out of over thirty which have been pathogenically associated with CMT. There should be no place for the indiscriminate use of multiple gene panels in the molecular diagnosis of disease.67
Conflict of interestsThe authors have no conflicts of interest to declare.
Please cite this article as: Berciano J, et al. Guía diagnóstica en el paciente con enfermedad de Charcot-Marie-Tooth. Neurología. 2012; 27:169–78.
