



La enfermedad de Charcot-Marie-Tooth (CMT) es la neuropatía hereditaria más frecuente. Clásicamente dividida según su patrón de herencia y de alteración de la velocidad de conducción motora (VCM) del nervio mediano, CMT incluye cinco grandes categorías: CMT1 (herencia autosómica dominante [AD] o ligada al sexo, y VCM < 38 m/s); CMT2 (herencia AD o ligada al sexo y VCM > 38 m/s); CMT4 (herencia autosómica recesiva [AR] y VCM muy lentificada); AR-CMT2 (forma recesiva con VCM > 38 m/s), y DI-CMT (forma intermedia con herencia AD y VCM entre 30 y 40 m/s). Pese a su estereotipado cuadro clínico (básicamente, semiología polineuropática sensitivo-motora y pie cavo), CMT ha resultado ser uno de los síndromes neurodegenerativos genéticamente más complejos, con 31 genes patogénicos clonados.
DesarrolloEl objetivo de esta guía es efectuar una revisión nosológica de la enfermedad de CMT, con énfasis en las directrices para llevar a cabo el diagnóstico molecular. A tal fin, revisamos los estudios de epidemiología y genética, y los genotipos descritos en España.
ConclusionesEn la inmensa mayoría de los pacientes con CMT, las mutaciones recaen en un reducido número de genes: para CMT1, PMP22, GJB1 y MPZ; para CMT2, MFN2 y GJB1; para CMT4, GDAP1, y NDRG1, HK1 y SH3TC2 (sujetos de etnia gitana); para AR-CMT2, GDAP1, y para DI-CMT, GJB1 y MPZ. Por su baja prevalencia, las mutaciones en otros genes sólo deberían investigarse cuando las anteriores han sido descartadas. Se desaconseja el uso indiscriminado de paneles de múltiples genes para el diagnóstico molecular de la enfermedad.
Charcot-Marie-Tooth disease (CMT) is the most frequent form of inherited neuropathy. In accordance with the inheritance pattern and degree of slowing of motor conduction velocity (MCV) of the median nerve, CMT encompasses five main forms: CMT1 (autosomal dominant [AD] or X-linked transmission and MCV < 38 m/s); CMT2 (AD or X-linked transmission and MCV > 38 m/s); CMT4 (autosomal recessive [AR] and severe slowing of MCV); AR-CMT2 (AR transmission and MCV > 38 m/s); and DI-CMT (intermediate form with AD transmission and MCV between 30 and 40 m/s). In spite of its stereotyped semiological repertoire (basically, symptoms and signs of sensory-motor polyneuropathy and pes cavus), CMT seems to be one of the most complex hereditary neurodegenerative syndromes, 31 causative genes having been cloned.
DevelopmentThis paper is aimed at performing a nosological review of the disease, emphasising the guidelines for its molecular diagnosis. Genetic epidemiological studies and genotypes reported in Spanish patients are revised.
ConclusionsIn the great majority of CMT cases, mutations involve a reduced number of genes, namely: for CMT1, PMP22, GJB1 and MPZ; for CMT2, MFN2 and GJB1; for CMT4, GDAP1, and NDRG1, HK1 and SH3TC2 (gypsies); for AR-CMT2, GDAP1; and for DI-CMT, GJB1 and MPZ. Given their low prevalence, mutations in other pathogenic genes should be investigated after discarding the previous ones. There is no place for the indiscriminate use of diagnostic CMT genetic panels.
La enfermedad de Charcot-Marie-Tooth (CMT) es la neuropatía hereditaria más frecuente con una prevalencia en España de 28,2 casos por 100.000 habitantes1. En lo esencial, se trata de un síndrome de inicio infantil o juvenil con semiología polineuropática motora y sensitiva, y pie cavo2–5. La CMT puede transmitirse con herencia autosómica dominante (AD), autosómica recesiva (AR) o ligada al cromosoma X. De acuerdo con los valores de conducción nerviosa hay formas desmielinizantes (velocidad de conducción motora [VCM] de nervio mediano < 38 m/s), axonales (VCM > 38 m/s) e intermedias (VCM 30-40 m/s)2–4. En buena correlación con las descripciones neurofisiológicas, estudios histológicos del sistema nervioso periférico (SNP) han demostrado un patrón dual, ya desmielinizante o axonal. Partiendo de estos datos clínicos, neurofisiológicos y patológicos, en la década de los setenta Dyck propuso una sencilla clasificación, unánimemente aceptada, que incluye los siguientes tipos: a) tipo I (CMT1, hipertrófico o desmielinizante) con herencia AD o AR; b) tipo II (CMT2, neuronal o axonal) con herencia AD o AR; c) tipo III (CMT3, usualmente con herencia AR) reservado para la enfermedad de Dejerine-Sottas o pacientes con formas graves de CMT hipomielinizante; d) formas ligadas al cromosoma X, e) formas complejas (p. ej., con atrofia óptica, sordera o degeneración pigmentaria de la retina)5. Aunque en la literatura la enfermedad ha sido también designada como neuropatía motora y sensitiva hereditaria (HMSN, en la abreviatura anglosajona que usaremos aquí por ser la que figura en OMIM y PubMed), en la actualidad se prefiere usar el acrónimo CMT. Merece la pena reseñar que el único signo clínico diferencial entre CMT1 y CMT2 es la presencia de engrosamiento, visible o palpable, de troncos nerviosos en CMT1.
Con los portentosos avances de la genética molecular en las dos últimas décadas, la nosología de la CMT ha estado en permanente cambio, al extremo de que la enfermedad incluye una treintena de genes patogénicos clonados (véase más adelante). Esta guía surge como una iniciativa del Programa 3 (Enfermedades Neuromusculares) de CIBERNED, siendo su objetivo hacer una breve revisión nosológica de la enfermedad y un análisis del diagnóstico de la enfermedad con énfasis en la selección de las pruebas moleculares.
Enfermedad de Charcot-Marie-Tooth en la era molecularAntes de empezar este apartado, conviene recordar que la CMT tiene una relación nosológica muy estrecha con otras dos formas de neuropatía hereditaria: neuronopatía motora hereditaria distal (dHMN) y neuropatía sensitiva y autonómica hereditaria (HSAN). Entre estos tres síndromes hay no solo solapamiento fenotípico, sino que se da el fenómeno de heterogeneidad alélica (idéntico fenotipo originado por diferentes mutaciones en el mismo gen y locus cromosómico) y de heterogeneidad de locus (mutaciones producidas en genes que se encuentran en diferentes loci cromosómicos dando lugar al mismo fenotipo). En aras de brevedad, sólo nos ocuparemos de la CMT haciendo puntual referencia a dHMN y HSAN allá donde convenga.
Mediante análisis de ligamiento genético se han localizado 37 loci con 31 genes clonados3,4,6–8. En HMN/HSAN se han descrito 12 loci adicionales con 9 genes clonados (para revisión véase la referencia 4). Se estima que, en su conjunto, todavía queda por descubrir la base molecular en un 33% de los casos de CMT9, lo cual es un reto que quizás se facilite con las nuevas técnicas de whole-genome sequencing10,11. Estos genes y sus respectivas proteínas constituyen un microarray de moléculas que son necesarias para el normal funcionamiento del SNP6. No deja de ser una ironía que la CMT, pese a la aparente simplicidad de su repertorio semiológico, haya resultado ser uno de los síndromes neurológicos genéticamente más complejos.
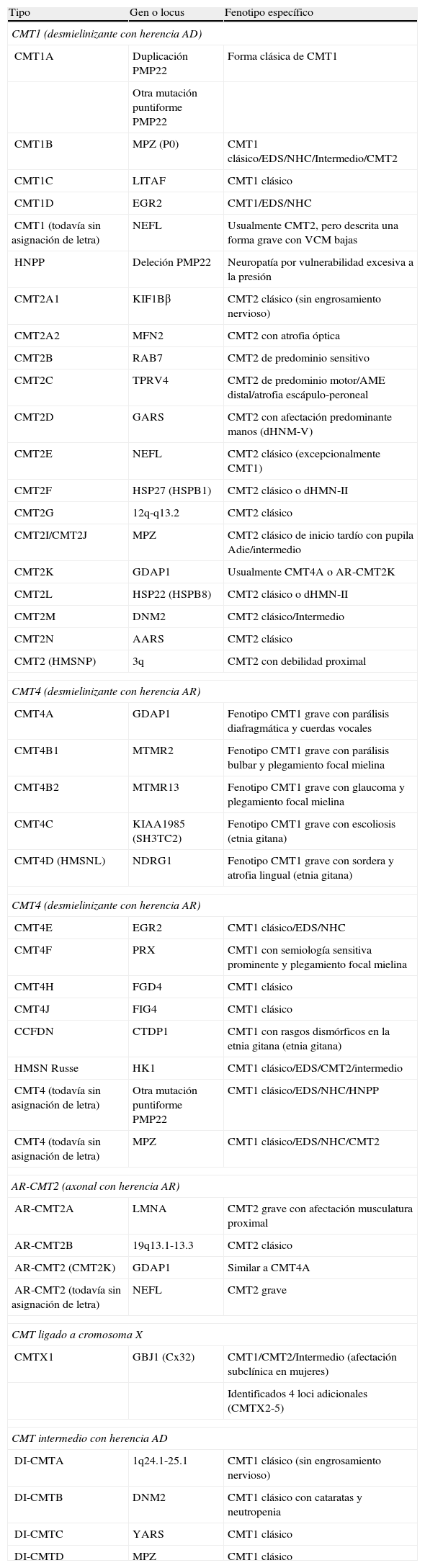
Adaptada de las referencias 4, 7, y 8, en la tabla 1 se recoge una clasificación clínico-genética actualizada de la CMT, que es provisional dado que no hay un criterio unánime en el uso de sus tipos y subtipos. Hay acuerdo universal en aceptar CMT1 como cabecera para los fenotipos desmielinizante con herencia AD. En CMT2, hay autores que incluyen formas axonales con herencia AD o AR, mientras que otros sólo incluyen las formas AD, creando el acrónimo AR-CMT2 para las formas axonales con transmisión AR; nosotros hemos seguido este criterio. El acrónimo CMT3 en la clasificación de Dyck5, aplicado para síndromes similares al descrito por Dejerine y Sottas12, desaparece y es sustituido por CMT4, donde se engloban todos los síndromes desmielinizantes con herencia AR. Se introduce, en fin, el acrónimo DI-CMT para las formas intermedias con transmisión AD.
Clasificación clínico-genética de CMT.
| Tipo | Gen o locus | Fenotipo específico |
| CMT1 (desmielinizante con herencia AD) | ||
| CMT1A | Duplicación PMP22 | Forma clásica de CMT1 |
| Otra mutación puntiforme PMP22 | ||
| CMT1B | MPZ (P0) | CMT1 clásico/EDS/NHC/Intermedio/CMT2 |
| CMT1C | LITAF | CMT1 clásico |
| CMT1D | EGR2 | CMT1/EDS/NHC |
| CMT1 (todavía sin asignación de letra) | NEFL | Usualmente CMT2, pero descrita una forma grave con VCM bajas |
| HNPP | Deleción PMP22 | Neuropatía por vulnerabilidad excesiva a la presión |
| CMT2A1 | KIF1Bβ | CMT2 clásico (sin engrosamiento nervioso) |
| CMT2A2 | MFN2 | CMT2 con atrofia óptica |
| CMT2B | RAB7 | CMT2 de predominio sensitivo |
| CMT2C | TPRV4 | CMT2 de predominio motor/AME distal/atrofia escápulo-peroneal |
| CMT2D | GARS | CMT2 con afectación predominante manos (dHNM-V) |
| CMT2E | NEFL | CMT2 clásico (excepcionalmente CMT1) |
| CMT2F | HSP27 (HSPB1) | CMT2 clásico o dHMN-II |
| CMT2G | 12q-q13.2 | CMT2 clásico |
| CMT2I/CMT2J | MPZ | CMT2 clásico de inicio tardío con pupila Adie/intermedio |
| CMT2K | GDAP1 | Usualmente CMT4A o AR-CMT2K |
| CMT2L | HSP22 (HSPB8) | CMT2 clásico o dHMN-II |
| CMT2M | DNM2 | CMT2 clásico/Intermedio |
| CMT2N | AARS | CMT2 clásico |
| CMT2 (HMSNP) | 3q | CMT2 con debilidad proximal |
| CMT4 (desmielinizante con herencia AR) | ||
| CMT4A | GDAP1 | Fenotipo CMT1 grave con parálisis diafragmática y cuerdas vocales |
| CMT4B1 | MTMR2 | Fenotipo CMT1 grave con parálisis bulbar y plegamiento focal mielina |
| CMT4B2 | MTMR13 | Fenotipo CMT1 grave con glaucoma y plegamiento focal mielina |
| CMT4C | KIAA1985 (SH3TC2) | Fenotipo CMT1 grave con escoliosis (etnia gitana) |
| CMT4D (HMSNL) | NDRG1 | Fenotipo CMT1 grave con sordera y atrofia lingual (etnia gitana) |
| CMT4 (desmielinizante con herencia AR) | ||
| CMT4E | EGR2 | CMT1 clásico/EDS/NHC |
| CMT4F | PRX | CMT1 con semiología sensitiva prominente y plegamiento focal mielina |
| CMT4H | FGD4 | CMT1 clásico |
| CMT4J | FIG4 | CMT1 clásico |
| CCFDN | CTDP1 | CMT1 con rasgos dismórficos en la etnia gitana (etnia gitana) |
| HMSN Russe | HK1 | CMT1 clásico/EDS/CMT2/intermedio |
| CMT4 (todavía sin asignación de letra) | Otra mutación puntiforme PMP22 | CMT1 clásico/EDS/NHC/HNPP |
| CMT4 (todavía sin asignación de letra) | MPZ | CMT1 clásico/EDS/NHC/CMT2 |
| AR-CMT2 (axonal con herencia AR) | ||
| AR-CMT2A | LMNA | CMT2 grave con afectación musculatura proximal |
| AR-CMT2B | 19q13.1-13.3 | CMT2 clásico |
| AR-CMT2 (CMT2K) | GDAP1 | Similar a CMT4A |
| AR-CMT2 (todavía sin asignación de letra) | NEFL | CMT2 grave |
| CMT ligado a cromosoma X | ||
| CMTX1 | GBJ1 (Cx32) | CMT1/CMT2/Intermedio (afectación subclínica en mujeres) |
| Identificados 4 loci adicionales (CMTX2-5) | ||
| CMT intermedio con herencia AD | ||
| DI-CMTA | 1q24.1-25.1 | CMT1 clásico (sin engrosamiento nervioso) |
| DI-CMTB | DNM2 | CMT1 clásico con cataratas y neutropenia |
| DI-CMTC | YARS | CMT1 clásico |
| DI-CMTD | MPZ | CMT1 clásico |
AARS: alanyl tRNA synthtase; AME: atrofia muscular espinal; CMT: enfermedad de Charcot-Marie-Tooth; CTDP1: CTD phosphatase subunit 1; DNM2: dynamin 2; EDS: enfermedad de Dejerine-Sottas; EGR2: early growth response 2; FDG4: RhoGEF; FIG4: Ptdlns(3,5)P25-phosphatase; GARS: glycyl tRNA synthetase; GBJ1: gap junction protein beta 1; GDAP1: ganglioside induced differentation associated protein 1; HK1: hexoskinase 1; HMSNL: hereditary motor and sensory neuropathy Lom; HNPP: hereditary susceptibility pressure palsy; HSP22: heat shock 22 kDa protein; HSP27: heat shock 27 kDa protein; KIF1Bβ: kinesin family member 1-Bβ; LITAF: lipopolysaccharide induced tumour necrosis factor; LMNA: lamin A/C; MFN2: mitofusin 2; MTMR2: myotubularin related protein 2; MTMR13: myotubularin related protein 13; NDRG1: N-myc downstream regulated gene; NEFL: neurofilament light polypeptide 68 kDa; NHC: neuropatía hipomielinizante congénita; PMP22: peripheral myelin protein 22; P0: myelin protein zero; PRX: periaxin; RAB7=RAB7, member RAS encogen family; SH3TC2: SH3 domain and tetratricopeptide repeats; TRPV4: transient receptor potencial vallinoid 4; YARS: tyrosyl tRNA synthetase. Véase también en http://neuromuscular.wustl.edu/time/hmsn.html.
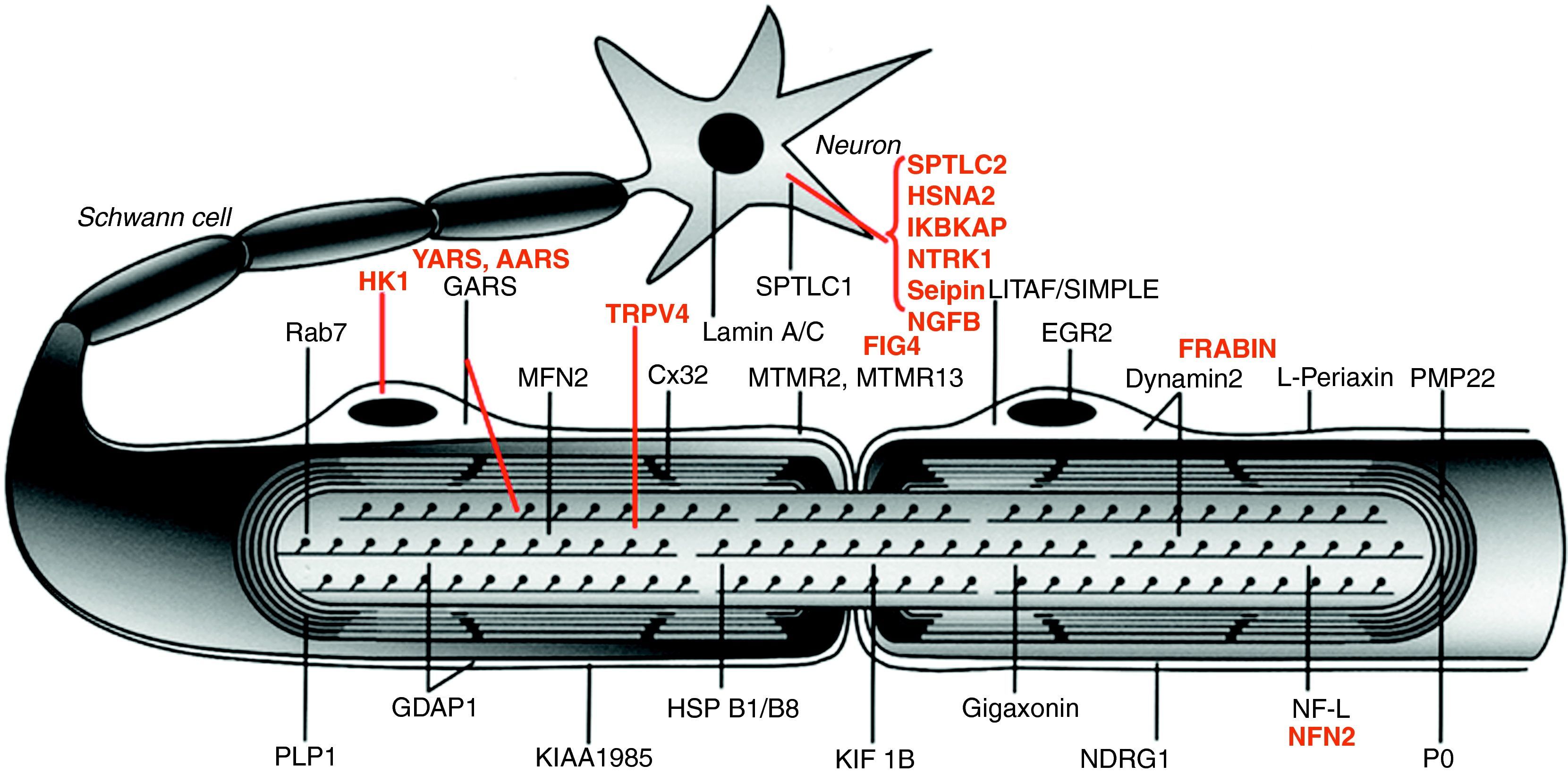
La figura 1 ilustra la localización de las proteínas mutadas, que era la predecible para aquellos componentes conocidos del SNP, tales como las proteínas PMP22 y MPZ (P0) de la mielina compacta. En otras situaciones, sin embargo, el descubrimiento de la proteína mutada patogénica resultó ser inesperado, como por ejemplo lo ilustra el caso de GDAP1, cuya función en el SNP se desconocía hasta la identificación de CMT4A13,14.
, HSN2 (hereditary sensory neuropathy type 2), NTRK1 (neurotrophic tyrosine kinase receptor type 1), IKBKAP (inhibitor of kappa light poypeptide gene enhancer in B-cells) y NGF1 (nerve growth factor beta polypeptide) están involucradas en la etiopatogenia de la neuropatías sensitivas y autonómicas hereditarias, no revisadas en este trabajo (véase texto). Adaptado de Niemann et al6.")
Dibujo esquemático de una fibra nerviosa mielinizada. Las proteínas mutadas, causales de CMT, HMN o HSAN, identificadas hasta 2006 figuran en negro, mientras que las descritas con posterioridad aparecen en rojo. Se mantienen las designaciones y acrónimos anglosajones porque son los que figuran en PubMed y OMIM. El significado de los acrónimos se recoge en el pie de la tabla 1. Nótese que mutaciones de SPTLC1 (serine palmitoyltransferase long chain base subunit 1), HSN2 (hereditary sensory neuropathy type 2), NTRK1 (neurotrophic tyrosine kinase receptor type 1), IKBKAP (inhibitor of kappa light poypeptide gene enhancer in B-cells) y NGF1 (nerve growth factor beta polypeptide) están involucradas en la etiopatogenia de la neuropatías sensitivas y autonómicas hereditarias, no revisadas en este trabajo (véase texto). Adaptado de Niemann et al6.
Desde un punto de vista didáctico y de acuerdo con Niemann et al6, los mecanismos etiopatogénicos de las proteínas mutadas se resumen del siguiente modo: a) por alteración del desarrollo y mantenimiento de la mielina; b) por alteración de la biosíntesis y degradación de proteínas; c) por alteración de la endocitosis y dinámica de membranas incluyendo la mitocondrial; d) por alteración del citoesqueleto axonal; e) seipinopatías15, y f) canalopatías por mutación de TRPV416. Revisaremos sucintamente estos seis apartados.
En las formas de CMT1/CMT4 por mutación de ciertos componentes de la mielina, se asume que el defecto de la célula de Schwann causa des/dismielinización con axonopatía axonal secundaria, que a la postre es responsable de la semiología clínica17–20. El síndrome más frecuente en este apartado es CMT1A, que representa el 55% de todos los casos de CMT y el 66,8% de CMT19 y es usualmente causado por una trisomía alélica de 17p11.2 de 1,5 Mb que contiene el gen PMP2221,22. Tal trisomía origina un exceso de dosis génica, lo cual implica una sobreproducción de PMP22 y su acumulación en la célula de Schwann, induciendo estrés de su retículo endoplásmico, que resulta en muerte celular programada. La deleción actúa reduciendo la expresión de PMP22, lo cual origina una mielina inestable que se manifiesta con un síndrome de neuropatía por vulnerabilidad excesiva a la presión (HNPP en el acrónimo anglosajón; véase tabla 1). En un pequeño porcentaje de casos duplicación/deleción pueden ocurrir como un fenómeno de novo. Otras mutaciones puntiformes (p. ej., sustituciones de una única base) del gen PMP22 son excepcionales y causan fenotipos graves, ya AD (probablemente por un mecanismo de ganancia de función) o AR (pérdida de función por fallo en la síntesis de PMP22)23. La proteína MPZ es cuantitativamente la más abundante de la mielina compacta y un elemento esencial para su compactación. En un 10% de los casos, la CMT es causada por mutaciones puntiformes de MPZ que resultan, ya en un fenotipo desmielinizante de inicio precoz AD (CMT1B) y excepcionalmente AR, o bien en fenotipos axonales de inicio tardío (CMT2I y CMT2J)24,25. Así, pues, la patología molecular de PMP22/MPZ ha desvelado que sus mutaciones pueden heredarse a través de transmisión tanto AD como AR, y que en el caso de MPZ sus mutaciones causan tanto un fenotipo desmielinizante como axonal, lo cual no hace sino subrayar que en el SNP el diálogo entre las células de Schwann y los axones acompañantes es continuo6. Tales fenómenos son aplicables a mutaciones causales de CMT en otros genes (véase tabla 1). GBJ1 (Cx32) es una proteína tipo gap de la mielina paranodal, cuyo gen está localizado en el cromosoma X. Segunda causa en frecuencia de CMT, mutaciones puntiformes en el gen GBJ1 originan una disfunción del tránsito radial de pequeñas moléculas entre la célula de Schwann y el axón6,26,27. Probablemente por un mecanismo de haploinsuficiencia, tales mutaciones causan un fenotipo más grave en varones que en mujeres, que neurofisiológicamente puede ser desmielinizante, intermedio o axonal. Otras causas más raras de CMT1/CMT4 incluyen mutación de EGR2 (gen que codifica para un factor transcripción involucrado en la regulación de genes de la mielina) y mutación de PRX (gen que codifica para una proteína de anclaje del citoesqueleto de la célula de Schwann).
La correcta composición y mantenimiento de los compartimentos membranáceos de las células de Schwann y las neuronas del SNP dependen de un perfecto equilibrio entre la síntesis de componentes estructurales y de señalización, y sus procesos de degradación6. Entre las proteínas mutadas implicadas en los procesos de endocitosis se cuentan las siguientes (tabla 1): a) fosfatasas (MTMR2, MTMR13 y FIG4), que causan fenotipos AR graves (CMT4B1, CMT4B2 y CMT4J) con plegamientos focales de la mielina (CMT4B1 y CMT4B2), b) GTPasas: DNM2 con fenotipo AD que puede ser tanto intermedio (DI-CMTB) como axonal28,29, RAB7 que causa CMT2B (un fenotipo similar al de HSNA1), y FRABIN que se asocia a CMT4H. Mutaciones NDRG1, un gen regulador de función poco conocida, causan un grave síndrome (CMT4D) en sujetos de etnia gitana. Por lo que respecta a componentes implicados en la síntesis, la clasificación y la degradación de las proteínas, las mutaciones afectan a los siguientes componentes: a) LITAF/SIMPLE, una ligasa de ubicuitina, que causa CMT1C, y b) GARS, YARS y AARS, proteínas implicadas en la carga del ARNt con glicina, tirosina y alanina, que originan CMT2D/dHMN-V, DI-CMTC, CMT2N, respectivamente (véase tabla 1).
Las neuronas del SNP, tanto sensitivas como motoras, deben mover proteínas, vesículas y organelas, por los largos trechos axonales que van desde el soma hasta sus terminales, lo cual requiere de un sistema de transporte complejo y eficiente. No puede sorprender el creciente número de formas de CMT axonal causadas por mutaciones de proteínas relacionadas con el citoesqueleto y el transporte de proteínas, vesículas y organelas (tabla 1)30. Mutaciones en la cadena ligera de los neurofilamentos (NEFL) ocasionan CMT2E y, excepcionalmente, CMT1F. Las proteínas de choque térmico (HSP) son macromoléculas ubicuas que en el SNP controlan el ensamblaje de los neurofilamentos. Mutaciones del gen HSP27 causan CMT2F/dHMN-II, mientras que mutaciones en HSP22 se asocian a CMT2L/dHMN-II. Recientemente, en una estirpe de CMT asociada a la mutación HSP27 R127W, con 10 pacientes explorados clínica y neurofisiológicamente, había casos con fenotipo de CMT2 y otros con fenotipo de HMN, lo cual no hace sino subrayar que ambos síndromes pueden ser una y única entidad nosólogica31. Las kinesinas son una familia de proteínas motoras que median el transporte axonal anterógrado sobre los microtúbulos, mientras que las dineínas median el transporte retrógrado. Mutaciones de KIF1Bβ se asocian a CMT2A1 y mutaciones de RAB7, que codifica para una GTPasa reguladora la función de dineínas, causa CMT2B. La morfología mitocondrial es determinada por un equilibrio entre procesos de fusión y fisión de la organela32. MNF2 es una GTPasa de la pared externa de la mitocondria, donde actúa como regulador de la fusión mitocondrial. Mutaciones puntiformes en el gen MFN2 causan CMT2A2, siendo actualmente la forma más frecuente de CMT2 (20%), con una quinta parte de los casos presentándose como mutaciones de novo33–35. Remedando HMSN-VI, en CMT2A2 puede haber atrofia óptica, especialmente en formas graves de inicio precoz. GDAP1 es la contrapartida de MFN2 participando en procesos de fisión mitocondrial. Mutaciones homocigotas de GDAP1 causan ya CMT4A o bien AR-CMT213,14,36,37; excepcionalmente, ciertas mutaciones en este gen causan enfermedad en estado heterocigoto (CMT2K)38,39. El gen LMNA codifica para una proteína de la membrana nuclear cuya mutación se asocia a AR-CMT2A; tiene interés señalar que mutaciones en el mismo gen pueden causar la miopatía de Emery-Dreifuss. El gen KIAA1985/SH3TC2 codifica para una proteína adaptadora y sus mutaciones causan un fenotipo grave (CMT4C)10 (véase tabla 1).
BSCL2 es un acrónimo derivado de Berardinelli-Seip congenital lipodystropy 2, un síndrome originalmente descrito en estirpes con lipoatrofia, resistencia a la insulina, hipertrigliciridemia, retraso mental y herencia AD. BSCL2 o Seipin es una proteína glucosilada del retículo endoplásmico, cuyas mutaciones activan la vía UPR (unfolded protein response) induciendo estrés del retículo endoplásmico y muerte celular programada15. Las seipinopatías están consideradas un nuevo modelo de enfermedad por alteración de la conformación proteica. Mutaciones puntiformes de BSCL2 causan un continuo de síndromes neurodegenerativos con transmisión AD, que incluyen dHMN-V, síndrome de Silver (paraparesia espástica y amiotrofia de manos), CMT2 y paraparesia espástica hereditaria; en un estimable porcentaje de casos, la mutación tiene penetrancia incompleta40.
TRPV4 es un miembro de canales catiónicos no selectivos implicados en la detección de estímulos físicos y químicos, y en múltiples funciones fisiológicas41. Mutaciones heterocigotas de TRPV4 se habían asociado a displasias óseas. Por análisis de ligamiento genético se sabía que CMT2C, la forma escápulo-peroneal de la atrofia muscular espinal (AME) y la forma congénita distal de AME podían ser síndromes alélicos (12q21-q24). Recientes estudios han demostrado que, en efecto, tales síndromes, a veces con penetrancia incompleta, se asocian a diversas mutaciones puntuales heterocigóticas en el dominio ankirina de TRPV416,42–45. Se desconoce el mecanismo por el que tales mutaciones causan degeneración del SNP. En todo caso, la enfermedad es un ejemplo prototípico de expresividad variable inter e intrafamiliar16,45.
Diagnóstico clínico de enfermedad de Charcot-Marie-ToothEl primer paso es establecer si el paciente presenta una neuropatía hereditaria. La respuesta puede ser evidente cuando la encuesta familiar demuestra que en la estirpe hay ancestros afectados, lo cual sugiere una herencia AD o ligada al sexo (cuando no hay transmisión varón-varón). La ocurrencia de enfermedad entre hermanos y la consanguinidad paterna sugiere una herencia AR. A veces, sin embargo, la encuesta familiar es negativa, en cuyo caso hay una serie de factores que orientan a una neuropatía genética, a saber: a) presentación en la infancia; b) curso clínico prolongado y lentamente progresivo; c) presencia de pie cavo, y d) a diferencia de las neuropatías adquiridas, ausencia de síntomas sensitivos positivos (parestesias o disestesias) pese a que haya clara semiología de déficit sensitivo8. Dado que a menudo los sujetos afectados tienen síntomas sutiles o incluso están asintomáticos, junto al probando es importante explorar el máximo número posible de sujetos en riesgo de la estirpe (casos secundarios). Esto posibilita detectar signos mínimos de enfermedad (p. ej., pie cavo o arreflexia) en casos subclínicos y, de este modo, perfilar mejor el patrón de herencia46. El examen de la musculatura de las extremidades inferiores mediante resonancia magnética ha ayudado a detectar signos incipientes de atrofia grasa en la musculatura intrínseca de los pies39,47,48.
El paso siguiente es el examen neurofisiológico que debe incluir la determinación de la VCM y VCS en al menos tres nervios. Se recomienda seguir las directrices requeridas por el Charcot-Marie-Tooth Neuropathy Score (CMTNS)49. A la hora de interpretar el grado de lentitud de la VCM, deberá tomarse en consideración la amplitud de los potenciales de acción motores compuestos (PAMC), porque una acusada caída de la amplitud del PAMC distal implica pérdida de fibras gruesas dependiente de la distancia, que puede llevar aparejada una reducción proporcional de la VCM. Para discernir entre caída de VCM por axonopatía o mielinopatía, se recomienda estudiar segmentos proximales del nervio, donde la conducción estará similarmente lentificada en casos de CMT desmielinizante y menos lentificada e incluso preservada en casos de CMT axonal17–19,50. En CMT1/CMT4 la lentificación de la VCM/VCS es difusa y uniforme, y la morfología de los PAMC y el índice de latencia terminal suelen estar preservados, lo cual está en contraposición con lo que acontece en las neuropatías inflamatorias adquiridas19. En las formas intermedias la VCM se sitúa entre 30 y 40 m/s (o 25 y 45, según algunos autores), tanto en troncos nerviosos con PAMC reducidos (usualmente segmentos distales) como preservados (usualmente segmentos proximales), es decir, la mutación génica causal actúa originando una disfunción tanto del axón como de la célula de Schwann51.
Actualmente, la biopsia de nervio queda reservada para casos en los que se plantean problemas de diagnóstico diferencial con otras neuropatías hereditarias (p. ej., amiloidosis) o con neuropatías adquiridas.
Diagnóstico molecular de enfermedad de Charcot-Marie-ToothDe la treintena de genes patogénicos hasta ahora identificados (tabla 1), en España sólo una pequeña y variable proporción de éstos está disponible para su estudio en laboratorios de genética molecular, sean de hospitales públicos o de entidades privadas. El gasto del diagnóstico molecular en nuestro medio está cubierto por el Ministerio de Sanidad y Consumo a través del Sistema de Información del Fondo de Cohesión (SIFCO). Ante el enorme y creciente número de genes patogénicos en la CMT y el estimable coste de los estudios moleculares, salta a la vista la imperiosa necesidad de dirigir las pruebas genéticas que se deben realizar. En una reciente comunicación de la Academia Americana de Neurología, se recomienda que el diagnóstico molecular de CMT se lleve a cabo basándose en el fenotipo clínico, el patrón de herencia y los hallazgos neurofisiológicos, empezando por el análisis de la duplicación/deleción de PMP22 cuando se trata de fenotipos desmielinizantes con herencia AD, o de mutaciones de GJB1 o de MFN2 cuando el fenotipo es axonal con herencia vertical52. Dada la enorme complejidad molecular de CMT, los estudios de la epidemiología genética son escasos9,53,54. Por ser el más reciente, extenso y detallado, nos ocuparemos a continuación del trabajo de Saporta et al9.
Los autores revisaron los casos remitidos a su consulta monográfica de CMT entre 1997 y 20079. Su casuística comprendía 1.024 pacientes, de los cuales 787 fueron diagnosticados de CMT. Un total de 527 enfermos (67%) fueron catalogados genéticamente; los restantes 260 quedaron como CMT no tipificados. Entre los 527 casos genéticamente definidos, los subtipos más frecuentes fueron los siguientes: CMT1A (55%; duplicación PMP22), CMT1X (15,2%; mutación de GJB1), HNPP (9,1%; deleción PMP22), CMT1B (8,5%; mutación de MPZ) y CMT2A (4%; mutación de MFN2). Se detectaron otras mutaciones en 23 pacientes (4,4% del total de CMT genéticamente definido) de CMT1 o CMT2, distribuidos del siguiente modo: CMT1C (LITAF), 5 casos; CMT1D (EGR2), 1 caso; CMT1E (mutación puntiforme PMP22), 5 casos; CMT2D (GARS), 3 casos; CMT2E (NEFL), 4 casos, y CMT2K (GDAP1), 5 casos. En este estudio sólo un 1,8% de los pacientes con CMT1 se quedaron sin tipificación genética, mientras que para CMT2 el porcentaje de tales casos no tipificados era del 65%. Únicamente 7 pacientes (1,4%) se encuadraron dentro de las formas autosómicas recesivas desmielinizantes (CMT4) con la siguiente distribución: CMT4A (GDAP1), 1 caso; CMT4C (SH3TC2), 3 casos; CMT4F (PRX), 1 caso, y CMT4J (FIG4), 2 casos.
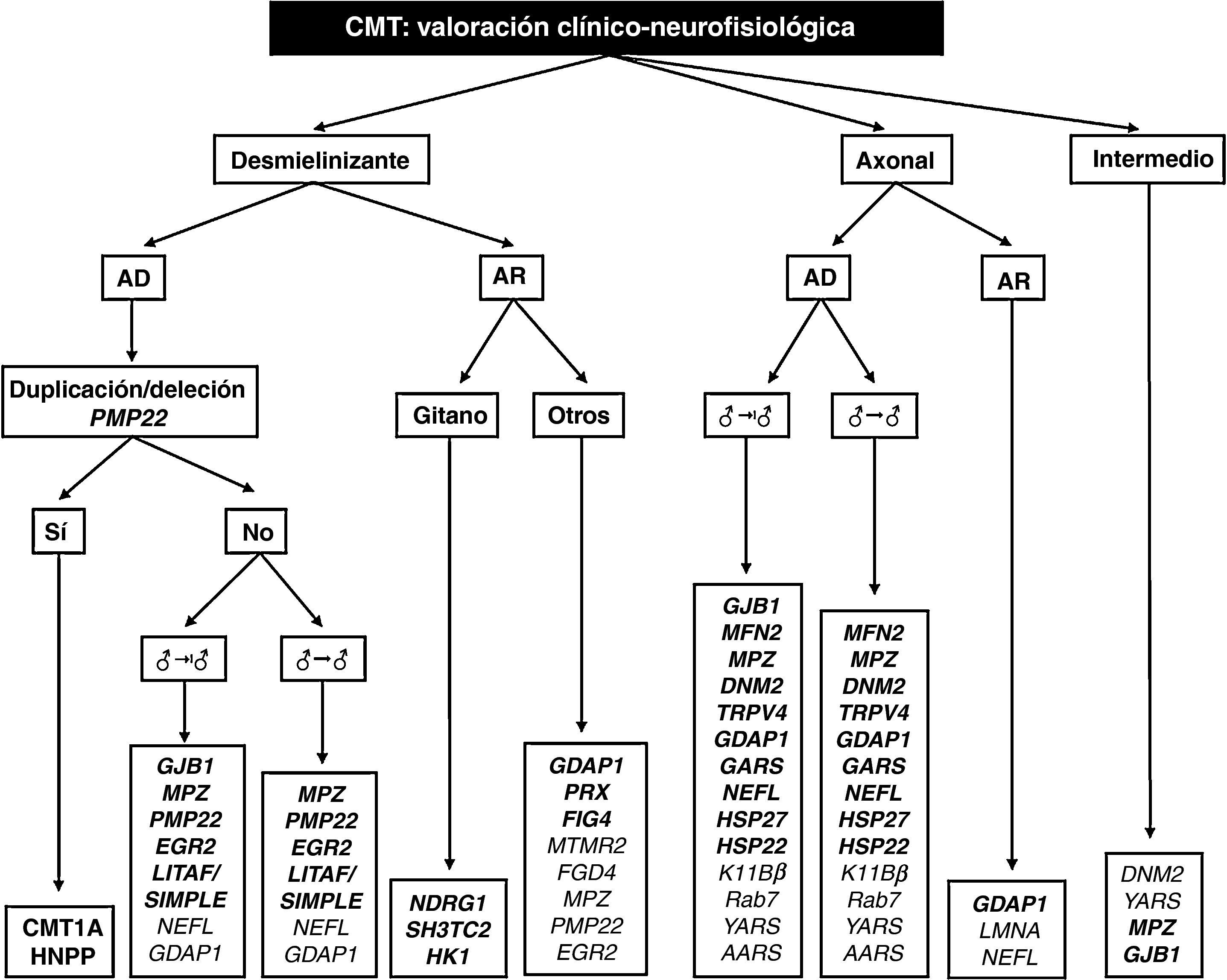
El trabajo de Saporta et al contribuye también a establecer los porcentajes de acierto para cada mutación estudiada, que varían entre el 80% para PMP22 y 13% para MFN29. Partiendo de ciertos rasgos fenotípicos y del estudio neurofisiológico, los autores proporcionan varios algoritmos para la elección del análisis molecular, que mejoran la fiabilidad del diagnóstico; sobre la base de éstos y de nuestra propia experiencia, hemos diseñado un algoritmo diagnóstico global para todas las formas de CMT (fig. 2). Comentaremos a continuación ciertas peculiaridades que facilitan el diagnóstico molecular en la CMT con transmisión vertical:
- –
Dado que el 89% de los pacientes con fenotipo clásico, sin retraso en el inicio de la marcha (< 15 meses), herencia AD y VCM en brazos entre 15 y 35 m/s tienen CMT1A, la primera y única prueba genética que se debe realizar ante un síndrome de CMT1 es la duplicación de PMP229. Si ésta es negativa, se procede a analizar GBJ1 y MPZ; las mutaciones en otros genes causales de CMT1 son excepcionales (véase antes y fig. 2).
- –
En pacientes con parecidas características a los del apartado anterior, pero con VCM muy lentificada (< 15 m/s), el estudio molecular puede empezar con PMP22 o MPZ, si bien el retraso del inicio de la marcha ocurre más a menudo en CMT1B que en CMT1A9,24.
- –
La mutación de GJB1 (CMT1X) puede asociarse con VCM dentro de los límites establecidos para CMT desmielinizante, CMT axonal o CMT intermedia. Por su alta prevalencia, las conexinopatías deberían ser consideradas en todo síndrome de CMT con aparente herencia vertical pero sin evidencia de transmisión varón-varón (véase fig. 2)9,27.
- –
El estudio molecular de enfermos con CMT axonal y herencia AD debería empezar con MFN2, GJB1 y MPZ. Característico de CMT2A2 de inicio precoz es la asociación con atrofia óptica (véase tabla 1)33. Un fenotipo de inicio tardío con pupila de Adie orienta a CMT2J25. Si bien la probabilidad de detectar mutaciones en los 11 genes restantes es escasa (fig. 2), se han descrito estirpes españolas de CMT2 con mutaciones en DNM2, GDAP1 y TRPV416,29,39,45; además, en nuestros laboratorios se han detectado casos inéditos y ocasionales de CMT2 asociado a mutaciones de NEFL, HSP27/HSPB1, y HSP22/HSPB22.
- –
La CMT con VCM intermedia y transmisión AD (DI-CMT en la tabla 1) incluye cuatro formas, la primera sin base molecular definida, y las otras tres ligadas a mutaciones de DNM2, YARS y MPZ. En nuestro medio, se han descrito fenotipos intermedios causados por mutaciones de MPZ55. Como hemos indicado antes, aquí cabe agregar mutaciones de GJB1. Merece la pena señalar que en la serie de Saporta et al las dos causas de DI-CMT fueron las mutaciones de GJB1 y MPZ9.
. AD: autosómico dominante; AR: autosómico recesivo; ♂→|♂: sin evidencia de transmisión varón-varón; ♂→♂: transmisión varón-varón.")
Algoritmo diagnóstico en el paciente con CMT. En negrita figuran mutaciones génicas descritas tanto en el trabajo de Saporta et al9 como en pacientes españoles (para detalles véase el texto, especialmente en relación con las prioridades del estudio molecular).
AD: autosómico dominante; AR: autosómico recesivo; ♂→|♂: sin evidencia de transmisión varón-varón; ♂→♂: transmisión varón-varón.
Las formas de CMT con transmisión AR representan en torno al 4% de la población europea con CMT, si bien en países con alta consanguinidad, como los del área mediterránea y Oriente medio, los porcentajes de AR-CMT pueden alcanzar entre el 30 y el 50% de todos los casos de CMT56. La información de la epidemiología genética de estas regiones es esencial para priorizar el estudio molecular de los 14 genes causales de las formas AR, ya desmielinizantes (CMT4) o axonales (AR-CMT2). De modo general (tabla 1), cabe decir que las formas recesivas tienen un inicio infantil, son más graves que los fenotipos dominantes, y que para las formas de CMT4 cursan con un acusado descenso de la VCM (usualmente, < 15 m/s)4. Los epónimos enfermedad de Dejerine Sottas y neuropatía hipomielinizante congénita probablemente sean aplicables para estas formas desmielinizantes recesivas57,58. En pacientes de etnia gitana, hay que considerar mutaciones fundadoras de NDRG1, SH3TC2 y HK1, las tres identificadas en España59–62. En nuestro medio, la mutación de GDAP1 es la causa más frecuente de CMT recesiva, sea CMT4A o AR-CMT2 (CMT2K)36,37. Se trata de un fenotipo grave de inicio infantil, con afectación de la musculatura proximal, y parálisis diafragmática y de las cuerdas vocales. Tiene interés señalar que mutaciones heterocigotas de GDAP1 pueden asociarse también con un fenotipo axonal menos grave y transmisión AD39. Idéntico fenómeno, de coparticipación de patrones de herencia AR y AD, ha sido excepcionalmente descrito para mutaciones de NEFL, HSP22 y MFN263–66. En la tabla 1 y la figura 2 se recogen otras mutaciones asociadas a CMT con transmisión AR.
ConclusiónEl diagnóstico de CMT debe partir de un adecuado estudio clínico, genealógico y neurofisiológico que permita establecer con cierta certidumbre el fenotipo de CMT (CMT1, CMT2, CMT4, AR-CMT2 o DI-CMT). Con este bagaje clínico y para la inmensa mayoría de los pacientes, el análisis genético puede dirigirse a la búsqueda de mutaciones un reducido grupo de genes, de la treintena de ellos patogénicamente relacionados con CMT. No hay lugar para el uso indiscriminado de paneles de múltiples genes en el diagnóstico molecular de la enfermedad67.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.

