


Hypoaldosteronism is an endocrine disease characterized by hyperkalemia and mild hyperchloremic metabolic acidosis with normal anion gap (type 4 renal tubular acidosis).
Causes of hypoaldosteronism include acquired disorders (hyporeninemic hypoaldosteronism, drug-induced angiotensin II inhibition, heparin therapy and primary adrenal insufficiency) and, less commonly, hereditary disorders. Adrenal aldosterone synthesis or renin release is affected in all these conditions.1
Aldosterone is a mineralocorticoid mainly acting in the kidney and, secondarily, in other organs (colon, lung, and sweat, lacrimal, and salivary glands). Aldosterone action, which requires a mineralocorticoid receptor and a sodium transporter protein, called sodium epithelial channel (SEC), regulates plasma sodium reabsorption and urinary potassium excretion.2 It is essential to differentiate decreased aldosterone production from aldosterone resistance.
The most commonly reported causes of the aldosterone resistance syndrome include treatment with potassium-sparing diuretics and antibiotic therapy with co-trimoxazole and pentamidine. A particularly uncommon condition is pseudohypoaldosteronism type 1 (PHA1).
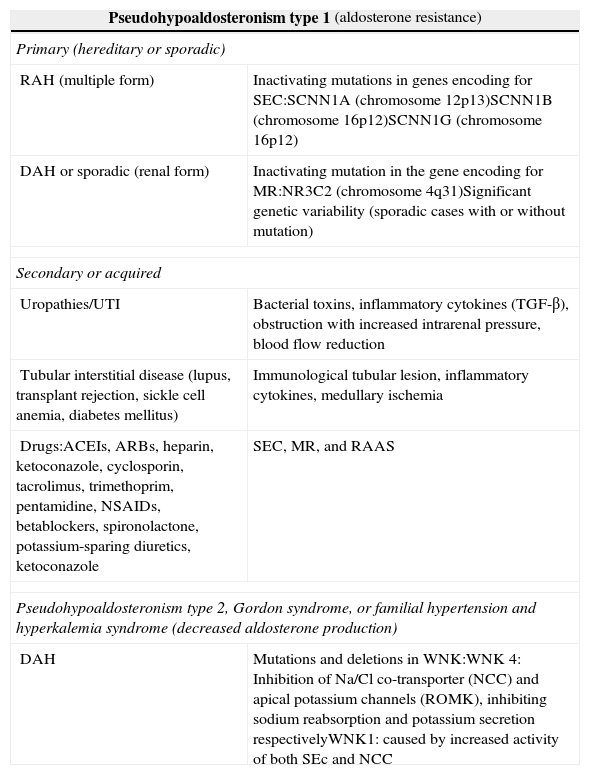
PHA1 may have a genetic basis and be inherited as a recessive autosomal disorder which affects SEC, impacting on all target organs (multiple form), or a dominant autosomal form, characterized by mutations in the gene encoding for the renal aldosterone receptor (renal form).3,4 Among secondary or acquired forms, special mention should be made of those derived from obstructive (organic or functional) and/or infectious uropathy, tubular interstitial disease, and side effects of drugs4 (Table 1).
Types of pseudohypoaldosteronism.
| Pseudohypoaldosteronism type 1 (aldosterone resistance) | |
| Primary (hereditary or sporadic) | |
| RAH (multiple form) | Inactivating mutations in genes encoding for SEC:SCNN1A (chromosome 12p13)SCNN1B (chromosome 16p12)SCNN1G (chromosome 16p12) |
| DAH or sporadic (renal form) | Inactivating mutation in the gene encoding for MR:NR3C2 (chromosome 4q31)Significant genetic variability (sporadic cases with or without mutation) |
| Secondary or acquired | |
| Uropathies/UTI | Bacterial toxins, inflammatory cytokines (TGF-β), obstruction with increased intrarenal pressure, blood flow reduction |
| Tubular interstitial disease (lupus, transplant rejection, sickle cell anemia, diabetes mellitus) | Immunological tubular lesion, inflammatory cytokines, medullary ischemia |
| Drugs:ACEIs, ARBs, heparin, ketoconazole, cyclosporin, tacrolimus, trimethoprim, pentamidine, NSAIDs, betablockers, spironolactone, potassium-sparing diuretics, ketoconazole | SEC, MR, and RAAS |
| Pseudohypoaldosteronism type 2, Gordon syndrome, or familial hypertension and hyperkalemia syndrome (decreased aldosterone production) | |
| DAH | Mutations and deletions in WNK:WNK 4: Inhibition of Na/Cl co-transporter (NCC) and apical potassium channels (ROMK), inhibiting sodium reabsorption and potassium secretion respectivelyWNK1: caused by increased activity of both SEc and NCC |
SEC: sodium epithelial channel; DAH: dominant autosomal heredity; RAH: recessive autosomal heredity; NCC: Na/Cl co-transporter; MR: mineralocorticoid receptor; ROMK: epithelial potassium channel; RAAS: renin–angiotensin–aldosterone system; WNK: with no lysine kinase.
PHA1 is characterized by aldosterone resistance, associated to hyponatremia, hypovolemia, hyperkalemia, and hyperchloremic metabolic acidosis. Plasma renin and aldosterone levels are markedly increased.
Although the syndrome has an insidious course, it may exceptionally lead to water and electrolyte emergencies. We therefore report the clinical case of a patient with severe dehydration, critical hyperkalemia, and urine output excessively high for the degree of dehydration. This was a 19-day-old male infant who was admitted to the pediatric ICU for dehydration and 19% weight loss (birth weight 3090g [10th–25th percentiles] after vaginal eutocic delivery at 41 weeks of pregnancy). The infant had not had clinical signs or symptoms of infection or fever.
Family history included grade 1 left vesicoureteral reflux (VUR), complicated with pyelonephritis at 15 days of life and requiring hospital admission, in a 6-year-old sister.
Laboratory tests showed leukocytosis (27,100WBC/mm3), with presence of band cells (9%). Chemistry showed greatly impaired renal and electrolyte profiles (urea, 234.9mg/dL; creatinine, 1.67mg/dL; sodium, 122.6mEq/L; chlorine, 90.4mEq/L; potassium, 11.25mEq/L; and calcium, 11.6mg/dL). Because of this critical potassium level, factitious hyperkalemia or preanalytical error (hemolyzed serum, EDTA-K3 contamination, excess compression or tourniquet time, and a drug-induced effect) were ruled out. Arterial blood gases showed metabolic acidosis (pH, 7.17; pCO2, 15mmHg; pO2, 105mmHg; HCO3−, 5.5mmol/L, and SBE, −20.1mmol/L). Analysis of urine collected by suprapubic puncture showed microscopic hematuria, pyuria, negative nitrites, proteinuria (150mg/dL), pH 6, specific gravity 1010, sodium 21mEq/L, and potassium 27.3mEq/L, with an osmolarity of 228mOsmol/kg. Blood, urine, and rectal swab samples for culture showed urinary tract infection (UTI) by E. coli susceptible to aminoglycosides, third-generation cefalosporins, and fosfonates. The patient is a rectal carrier of ESBL-producing K. pneumoniae, with negative blood cultures. Lumbar puncture provided no findings of interest, including cultures.
ECG showed characteristic signs of hyperkalemia (spiking T waves and PR in the upper limit of normal).
The etiological study was completed by abdominal ultrasonography, which showed bilateral ureterohydronephrosis, winding ureters, and hyperechogenic contents related to turbid urine. Based on these findings, voiding cystoureterography was performed, which ruled out structural obstruction and showed enlarged bladder, grade IV right and grade V left VUR, and incoordination of urinary detrusor/sphincter muscles consistent with functional obstruction, valve-like syndrome, uncoordinated voiding in the male infant, or Hinman–Allen syndrome.5 A renal scan with dimercaptosuccinic acid (DMSA) showed kidney function impairment (60% in the right and 40% in the left).
Adrenal function tests showed aldosterone levels higher than 2000pg/mL (17–130), plasma renin activity (PRA) of 27.9ng/mL/h (0.2–2.3), an aldosterone/PRA ratio higher than 72 (1.5–11). Free cortisol level in 24-h urine (16.7g/dL; 4.2–38.4) and ACTH (18pg/mL; 9–52), DHEA-S (375.2g/dL; 108–406), and 17-α-hydroxyprogesterone (1.46ng/mL; ≤4.5) levels were normal.
In conclusion, Hinman–Allen syndrome may result in a significant impact on kidney function, specifically on water and electrolyte homeostasis. The most important consequence is acquired PHA16 or renal aldosterone resistance syndrome. The main characteristics of PHA1 include hyponatremia, hyperkalemia and metabolic acidosis with abnormally high aldosterone levels. PHA1 has exceptionally been reported in children with obstructive uropathy or VUR and concomitant urinary infection. It has been postulated that PHA1 is caused by release of prostaglandins, thromboxane A2, leukotrienes, endothelin, angiotensin II, TNF-α, TGF-β1, and interleukins 1 and 6, associated to the renal parenchyma inflammatory process, mediated by bacterial endotoxins. As a result, vasoconstriction, decreased glomerular filtration rate, and natriuresis occur, related to transient damage to aldosterone receptors.7
During the first months of life, because of tubule immaturity, high aldosterone levels are required for adequate maintenance of water and electrolyte balance, which could be affected by uropathy, with or without an added infectious process.
Our patient had multiple risk factors involved in development of secondary PHA1, such as age, male sex, severe VUR, and UTI. The genetic basis of the condition was ruled out when hormone levels normalized after treatment. Otherwise, mutation in the NR3C2 gene (overlapping phenomenon) should have been evaluated.8
The initial symptomatic treatment consisted of rehydration, pH normalization, and correction of hyperkalemia (provision of sodium bicarbonate and calcium gluconate),9 together with implementation of a low pressure urinary system (urinary catheterization or vesicostomy), which reduces aggression of VUR on the upper urinary tract.10 Surgery was subsequently performed, consisting of bladder augmentation cystoplasty, which prevent progression to chronic renal failure.
Please cite this article as: Ruiz Ginés MÁ, Ruiz Ginés JA, Saura Montalbán J, Fontelles Alcover R, Piqueras Martínez AN. Pseudohipoaldosteronismo tipo 1 secundario a reflujo vesicoureteral: una urgencia endocrinológica. Endocrinol Nutr. 2014;61:495-497.
