





Current clinical practice guidelines recommend that a genetic study is considered in all patients diagnosed with pheochromocytoma or paraganglioma (PPGL). Our study objective was to know how many patients with PPGL undergo genetic studies at a non-specialized university hospital, the clinical factors involved in the decision to make the study, how many patients are found germline mutation, which are the affected genes, and what variables are related to presence of mutations.
Material and methodsAll patients diagnosed with PPGL at a tertiary university hospital from 2010 to 2015 were enrolled. Age and sex, tumor location and multiplicity, hormone secretion, presence of a clinical syndrome, family history, and medical department in charge were recorded and used to compare patients with (GEN+) and without (GEN−) genetic study, as well as patients with (MUT+) and without (MUT−) germline mutations.
ResultsThirty-nine patients were enrolled (21 females and 18 males with a mean age of 53.9±17.8 years). A genetic study was performed in 54% of patients with PPGL. These were younger, were more frequently seen by endocrinologists, and had more often a family history related to PPGL, multiple PPGLs, or hormonally functional tumors. Unilateral head and neck paragangliomas were less common. Germline mutations (3 RET, 3 SDHB, 1 SDHD) were found in 33% of patients, who were younger and more frequently had a clinical syndrome, multiple PPGLs. and a family history of PPGL.
ConclusionAlthough current clinical practice guidelines recommend that genetic studies are considered in all patients diagnosed with PPGL, studies was requested for 54% of such cases in our healthcare area. Predisposing germline mutations were found in 33% of studies.
Las actuales guías de práctica clínica recomiendan considerar el estudio genético en todos los pacientes con diagnóstico de feocromocitoma o paraganglioma (PPGL). El objetivo de nuestro trabajo es conocer el porcentaje de solicitud de estudio genético en el PPGL en un hospital universitario no especializado, los factores implicados en dicha solicitud, cuántos de ellos presentan mutación germinal, cuáles son los genes afectados y qué variables se relacionan con la presencia de mutaciones.
Material y métodosSe incluyó a todos los pacientes con PPGL diagnosticados en el área sanitaria de un hospital universitario de tercer nivel entre 2010 y 2015. Se recogieron las variables: edad, localización, único o múltiple, secreción hormonal, cuadro sindrómico, antecedentes familiares y servicio responsable. Se comparó a los pacientes con estudio genético realizado (GEN+) frente a aquellos no estudiados (GEN–), y pacientes con mutación (MUT+) frente a aquellos sin mutación predisponente (MUT–).
ResultadosSe incluyó a 39 pacientes (21 mujeres y 18 varones con edad media 53,9±17,8 años). Se hizo estudio genético al 54% y estos eran más jóvenes, con antecedentes familiares, múltiples, secretores y con mayor frecuencia vistos por Endocrinología. Hubo menos paragangliomas de cabeza y cuello unilaterales. Un 33% tenía mutación germinal (3 RET, 3 SDHB, un SDHD) y estos eran más jóvenes, más sindrómicos, múltiples o con antecedentes familiares.
ConclusionesAunque las guías de práctica clínica recomiendan considerar la realización de un estudio genético a todos los pacientes con PPGL, en nuestra Área de Salud se solicitó en el 54% de ellos. Un 33% de ellos presentaron mutación germinal predisponente.
Pheochromocytoma and paraganglioma (PPGL) are tumors derived from chromaffin tissue. Most of these tumors produce catecholamines which are responsible for the sympathomimetic symptoms of the disease, high blood pressure being the most common finding. Parasympathetic paragangliomas of the head and neck usually do not secrete catecholamines. They consequently do not cause arterial hypertension and are usually diagnosed as a result of their mass-generating effect; as incidentalomas; or during the follow-up of patients with some predisposing mutation.
Since 1990, at least 15 susceptibility genes for PPGL have been identified. Mutations in three of them are associated with syndromic conditions: von Hippel-Lindau (VHL), MEN2 (RET), and type 1 neurofibromatosis (NF1). Another 5 genes are implicated in the synthesis of the mitochondrial enzyme succinate dehydrogenase (SDH): two genes encode for its catalytic subunits (SDHA and SDHB); another two encode for the anchoring subunits (SDHD and SDHC); and the SDHAF2 gene encodes for the protein responsible for the flavinization of SDHA, needed for adequate enzyme assembly. Mutations of these genes are implicated fundamentally in the development of paragangliomas. Other susceptibility genes that have been described in a few cases include the TMEM127, MAX, FH, EGLN1/PHD2, KIF1β, IDH1 and HIF2α/EPAS1 genes.1–3 Pheochromocytomas and paragangliomas can be grouped into two clusters according to their genic expression profile. Cluster 1 comprises tumors with mutations in genes involved in the response to hypoxia (SDHx [cluster 1A] and VHL [cluster 1B]): their mutations activate these pathways and angiogenesis under conditions of normoxia. Cluster 2 in turn includes tumors with mutations in the RET, NF1, TMEM127 and MAX genes, all implicated in the activation of the MAPK and PI3K-AKT-mTOR intracellular signaling cascades, and in cell replication.2–4
The current guidelines1 recommend that all patients should receive information about their genetic diagnosis and should take part in decision-making regarding the convenience of establishing the diagnosis. However, no studies have examined the degree of compliance with this recommendation in actual clinical practice in different settings. The approach proposed by these guidelines is to conduct a sequential analysis of genes, starting with those most likely to have mutated, considering the clinical characteristics of each case and the relative frequency of the different genes, until a pathogenic mutation is identified. However, this approach is costly in terms of time and money, and several groups have reported excellent results using next generation sequencing (NGS) panels, which simultaneously study virtually all the genes known to predispose to PPGL, at a lower cost.5–7
In recent years, several working groups in the genetics of pheochromocytomas and paragangliomas have found the proportion of PPGL patients carrying a predisposing germline mutation to be between 14 and 41%.5,6,8–14 However, most of these studies come from reference centers that receive samples from a number of hospitals. We therefore cannot be certain that all patients diagnosed with PPGL are being studied. Genetic studies have possibly not been made in some patients with a low probability of germline mutation (single, non-syndromic adrenal gland tumors in older patients with no family history). Such studies are more likely in patients at greater risk, thereby falsely increasing the percentage of patients with mutations.
The present study was carried out to determine the proportion of PPGL patients seen in a non-specialized university hospital who are subjected to genetic study; the factors related to the request for such study; the proportion of patients that carry a germline mutation; the genes involved; and the variables related to the presence of mutations. As a secondary objective, we aimed to determine the proportion of PPGL patients monitored in departments corresponding to specialities other than Endocrinology, and to assess the consistency of the criteria used by such departments for requesting genetic study in such patients with respect to the criteria used in Endocrinology.
Material and methodsA review was made of the medical records of all patients with a diagnosis of PPGL reflected in the discharge or pathology reports between 1 January 2010 and 31 December 2015, at a third-level university hospital. We included all patients with histopathological confirmation of the diagnosis (38 subjects), as well as a female patient not subjected to surgery but with a clear diagnosis based on the clinical, biochemical, and radiological findings. Three patients (all with head and neck paraganglioma) underwent surgery at another hospital. Data collection from the coding of both discharge and pathology reports ensured the retrieval of virtually all the patients with this type of disorder.
The following variables were analyzed: patient age at diagnosis, gender, tumor location, multiplicity, hormone secretion (total and fractionated metanephrines in 24-hour urine as determined by high performance liquid chromatography [HPLC]), associated syndromic manifestations, family history of PPGL, the department in charge of the patient, the existence or not of a genetic study, and identified germline mutations. These variables were used to compare patients with (GEN+) and without genetic study (GEN−). Furthermore, among the GEN+patients, comparison was made of those with a mutation predisposing to PPGL (MUT+) versus those with no such mutation (MUT−). The variable “gender” was only used for the descriptive analysis of the study population, and the variable “hormone secretion” was not considered when comparing MUT+ versus MUT−.
The genetic study was made using peripheral blood samples. Most of the samples were sent to the Hereditary Endocrine Cancer Group of the Spanish National Cancer Research Center (Centro Nacional de Investigaciones Oncológicas [CNIO]) in Madrid (Spain). Up until 2014, Sanger and multiplex ligand-dependent probe amplification (MLPA)10,14–16 techniques were used to study the different genes on a sequential basis, starting with those most likely to have mutated, and taking the clinical characteristics of each case and the relative frequency of the different genes into consideration until a pathogenic mutation could be identified. The more recent cases were studied using NGS panels.7
The statistical analysis was performed using the G-Stat 2.0 package. Patient age was reported as the mean±standard deviation (SD), while the nonparametric Mann–Whitney U-test was used for populational inferences between subgroups, since all of them presented fewer than 30 cases. The other variables (all of a qualitative nature) were reported as proportions, and subgroups were compared using the chi-squared test or Fisher exact test (if any of the cells had fewer than 5 subjects).
The study was approved by the Clinical Research Ethics Committee of our hospital.
ResultsDescriptive analysis of the study populationThe study comprised a total of 39 patients (21 females [53.8%] and 18 males [46.5%]). The mean patient age at diagnosis was 53.9±17.8 years.
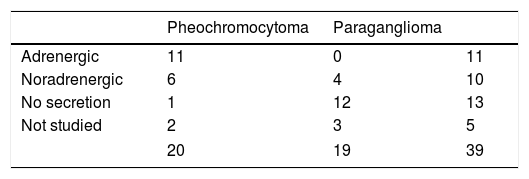
Eighteen patients (47%) presented an adrenal gland tumor, with one bilateral metachronous presentation corresponding to a patient with MEN2A syndrome. An additional 19 patients (48%) had paragangliomas: 13 of the head and neck (10 single, 1 recurrent and 2 bilateral presentations) and 6 thoracoabdominal tumors (one recurrent and one malignant presentation). Lastly, two patients developed paragangliomas some years after the surgical removal of a pheochromocytoma (Fig. 1).

Seventeen of the 20 pheochromocytomas (85%) were secretory (11 adrenergic and 6 noradrenergic). Of the remaining three cases, one was considered to be non-secretory. This was a 32-year-old male monitored for medullary thyroid carcinoma and carrying an RET gene mutation, in which a 1-cm adrenal incidentaloma indicative of pheochromocytoma was identified, with normal urinary metanephrine levels. The histological findings were consistent with pheochromocytoma. The corresponding information was not available in the remaining two patients, one of whom had been studied at another hospital. Of the 19 paragangliomas, four (21%) exhibited a noradrenergic secretory pattern, 12 were non-secretory, and three had not undergone preoperative metanephrine assessment because PPGL had not been initially suspected (Table 1).
Two patients reported a family history of paragangliomas and three reported a history of medullary thyroid carcinoma. The rest had no relevant family history, or at least no such history had been documented in the case history.
Only one of the 39 cases in our study exhibited malignant behavior. This was a woman diagnosed with an inter-aortocaval paraganglioma at 23 years of age who subsequently developed liver and bone metastases. The genetic studies conducted to date have revealed no germline mutations, though one somatic mutation in the NF1 gene has been identified.
In our series, 23 patients (59%) were seen in the Endocrinology department (15/18 pheochromocytomas, 2/2 paragangliomas+pheochromocytomas, 4/6 thoracoabdominopelvic paragangliomas and 2/13 head/neck paragangliomas), 9 in Vascular Surgery (23%) – all with single non-secretory head and neck paragangliomas – and the remaining 7 (18%) in the ENT, Nephrology, Internal Medicine, Urology and Neurosurgery departments.
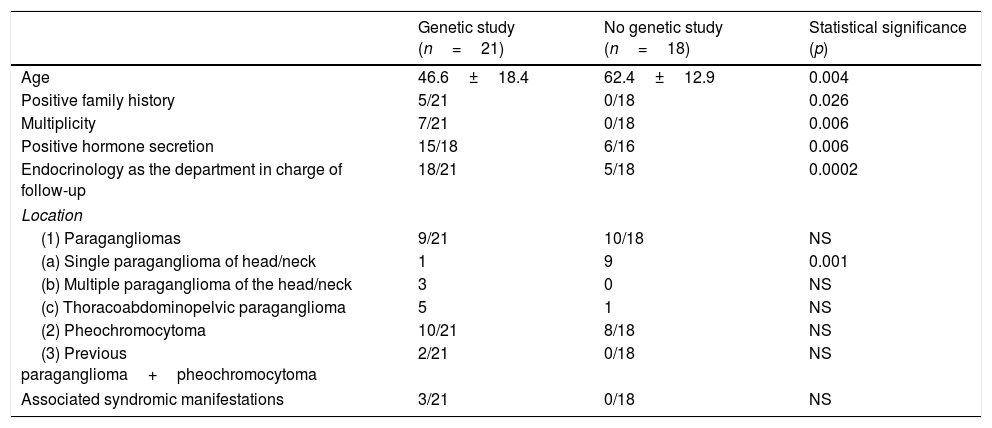
Which PPGL patients were subjected to genetic study and what distinguished them from those not studied?Germline mutation studies were carried out in 21 patients (GEN+ 54%), and these were compared with the patients not subjected to germline mutation study (GEN−). The studied patients were significantly younger, had more family antecedents, with tumors that were more often multiple (bilateral or recurrent) and secretory, and had been seen by Endocrinology. The tumors in these subjects were less frequently single head and neck lesions (Table 2). Genetic study was carried out in all syndromic patients, though the difference with respect to the patients not subjected to study failed to reach statistical significance, probably because of the small sample size involved.
Comparison of PPGL patients subjected to genetic study (n=21) versus those without genetic study (n=18).
| Genetic study (n=21) | No genetic study (n=18) | Statistical significance (p) | |
|---|---|---|---|
| Age | 46.6±18.4 | 62.4±12.9 | 0.004 |
| Positive family history | 5/21 | 0/18 | 0.026 |
| Multiplicity | 7/21 | 0/18 | 0.006 |
| Positive hormone secretion | 15/18 | 6/16 | 0.006 |
| Endocrinology as the department in charge of follow-up | 18/21 | 5/18 | 0.0002 |
| Location | |||
| (1) Paragangliomas | 9/21 | 10/18 | NS |
| (a) Single paraganglioma of head/neck | 1 | 9 | 0.001 |
| (b) Multiple paraganglioma of the head/neck | 3 | 0 | NS |
| (c) Thoracoabdominopelvic paraganglioma | 5 | 1 | NS |
| (2) Pheochromocytoma | 10/21 | 8/18 | NS |
| (3) Previous paraganglioma+pheochromocytoma | 2/21 | 0/18 | NS |
| Associated syndromic manifestations | 3/21 | 0/18 | NS |
NS: nonsignificant.
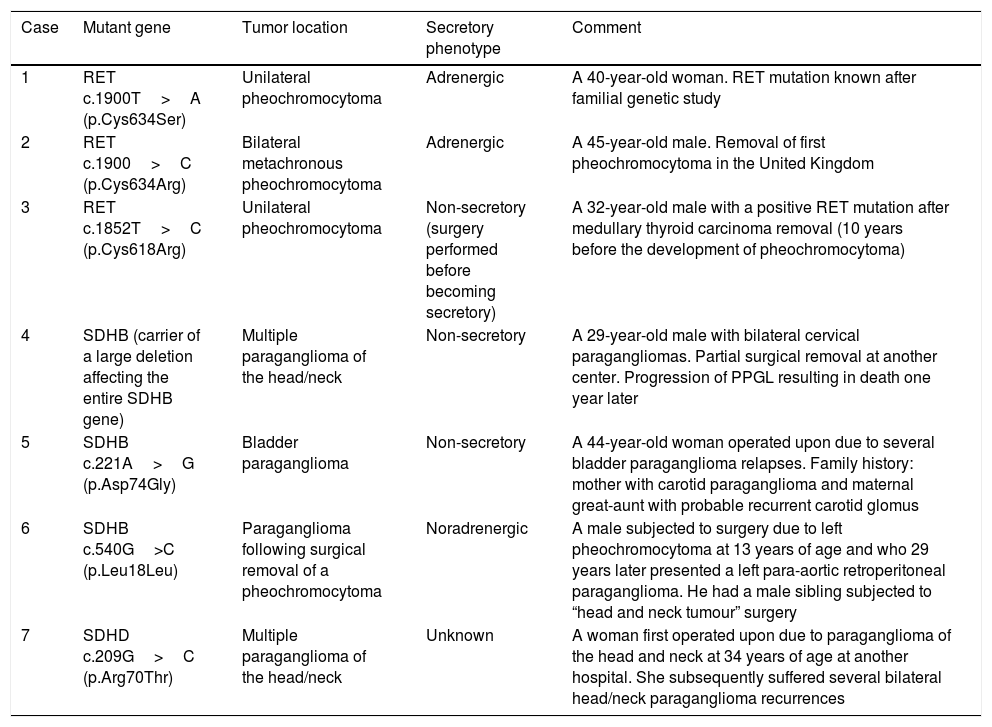
Of the 21 patients subjected to genetic study, 7 had germline mutations (33%): three in RET, three in SDHB and one in the SDHD gene. Their clinical characteristics are summarized in Table 3.
Clinical characteristics of the patients carrying germline mutations predisposing to PPGL.
| Case | Mutant gene | Tumor location | Secretory phenotype | Comment |
|---|---|---|---|---|
| 1 | RET c.1900T>A (p.Cys634Ser) | Unilateral pheochromocytoma | Adrenergic | A 40-year-old woman. RET mutation known after familial genetic study |
| 2 | RET c.1900>C (p.Cys634Arg) | Bilateral metachronous pheochromocytoma | Adrenergic | A 45-year-old male. Removal of first pheochromocytoma in the United Kingdom |
| 3 | RET c.1852T>C (p.Cys618Arg) | Unilateral pheochromocytoma | Non-secretory (surgery performed before becoming secretory) | A 32-year-old male with a positive RET mutation after medullary thyroid carcinoma removal (10 years before the development of pheochromocytoma) |
| 4 | SDHB (carrier of a large deletion affecting the entire SDHB gene) | Multiple paraganglioma of the head/neck | Non-secretory | A 29-year-old male with bilateral cervical paragangliomas. Partial surgical removal at another center. Progression of PPGL resulting in death one year later |
| 5 | SDHB c.221A>G (p.Asp74Gly) | Bladder paraganglioma | Non-secretory | A 44-year-old woman operated upon due to several bladder paraganglioma relapses. Family history: mother with carotid paraganglioma and maternal great-aunt with probable recurrent carotid glomus |
| 6 | SDHB c.540G>C (p.Leu18Leu) | Paraganglioma following surgical removal of a pheochromocytoma | Noradrenergic | A male subjected to surgery due to left pheochromocytoma at 13 years of age and who 29 years later presented a left para-aortic retroperitoneal paraganglioma. He had a male sibling subjected to “head and neck tumour” surgery |
| 7 | SDHD c.209G>C (p.Arg70Thr) | Multiple paraganglioma of the head/neck | Unknown | A woman first operated upon due to paraganglioma of the head and neck at 34 years of age at another hospital. She subsequently suffered several bilateral head/neck paraganglioma recurrences |
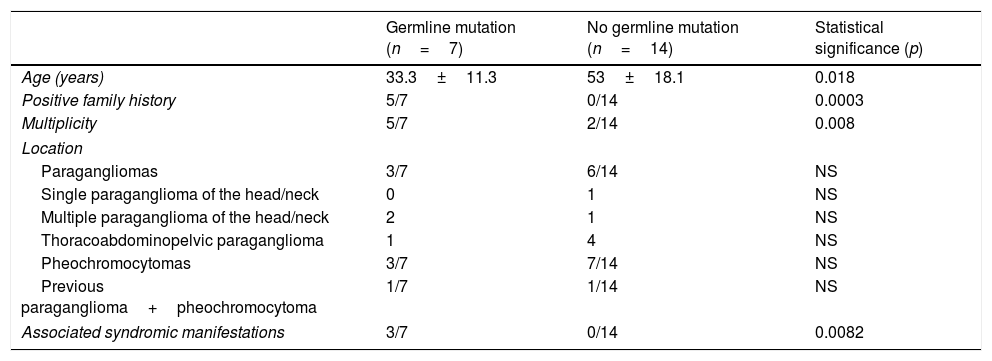
These 7 patients were significantly younger, more often presented syndromic conditions, and had more family antecedents and multiple tumors. There were no significant differences in tumor location between the patients with and without germline mutations. The corresponding data are reported in Table 4.
Comparison of patients with positive germline mutation study findings (n=7) versus patients without germline mutations (n=14).
| Germline mutation (n=7) | No germline mutation (n=14) | Statistical significance (p) | |
|---|---|---|---|
| Age (years) | 33.3±11.3 | 53±18.1 | 0.018 |
| Positive family history | 5/7 | 0/14 | 0.0003 |
| Multiplicity | 5/7 | 2/14 | 0.008 |
| Location | |||
| Paragangliomas | 3/7 | 6/14 | NS |
| Single paraganglioma of the head/neck | 0 | 1 | NS |
| Multiple paraganglioma of the head/neck | 2 | 1 | NS |
| Thoracoabdominopelvic paraganglioma | 1 | 4 | NS |
| Pheochromocytomas | 3/7 | 7/14 | NS |
| Previous paraganglioma+pheochromocytoma | 1/7 | 1/14 | NS |
| Associated syndromic manifestations | 3/7 | 0/14 | 0.0082 |
NS: nonsignificant.
Although the current clinical practice guidelines recommend germline mutation testing to be considered for all patients diagnosed with PPGL,1 the extent to which this recommendation is being implemented in the clinical setting is not known. The main objectives of the present study were firstly to determine how many patients diagnosed with pheochromocytoma or paraganglioma had been investigated for the presence of predisposing germline mutations, and secondly to establish what mutations were found in the studied patients. We decided to include patients diagnosed after the year 2010, because the main genes predisposing to PPGL were already known at that time; several research groups had confirmed the existence of a significant percentage of non-syndromic patients with no family history exhibiting mutations in these genes; and some experts had already recommended the study of these mutations in all or in most patients with PPGL.9,11,14 We did not collect data on the study of somatic mutations in tumor tissues, since the recommendations referring to such studies have remained vague in the clinical practice guidelines of recent years.
The study of predisposing germline mutations was conducted in 54% of the patients diagnosed with PPGL. There are no similar studies with which to compare this figure either in Spain or in other countries. However, the variability we have found between different specialties within the same hospital suggests that there is probably considerable variability among different clinical settings. Our center is a third-level university hospital not specializing in this type of disease. As such, it is probably representative of most Spanish hospitals in which PPGL patients are seen.
The probability of finding germline mutations increases if the patient is young; has a family history or tumors characteristic of PPGL-associated syndromes; and when PPGL is located outside the adrenal gland or proves bilateral, multiple or metastatic.2,3 We were able to confirm that these risk factors had been taken into account when requesting the genetic study in our setting, because the patients subjected to genetic testing were younger, had more syndromic conditions and a greater incidence of family antecedents, and more often presented multiple tumors. In fact, all the patients with a family history or with clinical characteristics of MEN2A syndrome had been studied. The only patient with malignant PPGL had also been investigated, without a germline mutation being found. However, an extra-adrenal tumor location was not associated with an increased request for genetic studies, particularly in single head and neck tumors. It should be noted that the probability of a genetic study being requested is largely dependent upon the department in charge of patient care.
The percentage of patients with mutations in our series (33%) was similar to that reported by other groups, and the distribution of the mutant genes was also similar to that described in other series.5,6,8–14 We possibly could have recorded a different percentage if all the patients had been studied. On the one hand, those not studied were older and had less syndromic disease, fewer family antecedents and fewer multiple tumors, making the presence of germline mutations less likely. On the other hand, however, 10 of the 18 patients not studied had extra-adrenal tumors, which are often associated with mutations.
The patients most likely to have a germline mutation are those presenting non-metastatic, non-syndromic unilateral adrenal pheochromocytoma without a family history, particularly if such individuals are over 45 years of age. In our series, a genetic study was carried out in 7 of the 17 patients with these characteristics – all over 45 years of age – and no germ mutations were found in any of them. Other research groups have reported mutation percentages ranging from 4-28% in these patients. In this respect, Cascón et al. found mutations in 3.9% of the non-syndromic patients with no family history and over 40 years of age,9 while Currás-Freixes et al. found mutations in 4.5% of all single, sporadic and non-syndromic pheochromocytomas.10 A somewhat higher percentage was reported by Jafri et al., who described mutations in 28% of the sporadic pheochromocytomas, though the mean age of the patients was 36 years, which is rather lower than in the other series.13
Among the limitations of our study, it should be mentioned that some patients may have rejected the genetic study without this fact being recorded in the case history, though this seems unlikely. In addition, the review of the case histories suggested that these were not always exhaustive in evaluating a family history (with guided questioning about PPGL in locations other than that of the patient and about other syndromic tumors). There was also variability in the number of genes analyzed in patients with negative genetic study findings. The use of classical sequencing in most patients, and of NGS techniques in the most recent patients, is unlikely to have influenced the overall results.
Our understanding of the molecular bases underlying the pathogenesis of pheochromocytoma and paraganglioma has grown enormously in recent years, and we can assume that in the near future the identification of the mutation in a given patient will contribute to the choice of the most adequate treatment and to establishing the prognosis. Our study reveals a clear variability among different departments regarding the likelihood of genetic studies being requested, with an unjustifiably low number of requests in the case of single paragangliomas of the head and neck. We therefore consider it necessary that patient management protocols based on a consensus among the different specialists involved be established.
AuthorshipInterventions: CMJ and MO performed data collection from the case histories. RPQ, CG, PP and CL were the physicians attending the patients in the Endocrinology department. CMJ and CL developed the original idea of the study, and were responsible for the drafting and the final correction of the manuscript.
Conflicts of interestThe authors state that they have no conflicts of interest. This work is original and has not been previously published. All the authors have approved the publication of the study.
Thanks are due to the Hereditary Endocrine Cancer Group of the Spanish National Cancer Research Center (Centro Nacional de Investigaciones Oncológicas [CNIO]) and in particular to María Currás and Mercedes Robledo for conducting the genetic studies.
Please cite this article as: Jiménez CM, Quílez RP, Gonzalvo C, Pinés P, Olmos M, Lamas C. Estudio de mutaciones germinales en pacientes con feocromocitomas y paragangliomas atendidos en un hospital universitario de tercer nivel: ¿qué pacientes se estudian y qué resultados se encuentran? Endocrinol Diabetes Nutr. 2018;65:508–514.
