








Introduction and aim. Hepatocyte growth factor (HGF) has been shown to ameliorate liver inflammation and fibrosis; however, the mechanism underlying its effects in non-alcoholic steatohepatitis (NASH) is unclear. This study aimed to analyse the relationship between the JAK2-STAT3 signalling pathway and the ameliorating effect of HGF on NASH.
Material and methods. Mice were fed a high-fat diet (HFD) for 16 weeks, and then plasma and hepatic tissues were collected. Histological and clinical chemistry assays were performed to assess liver disease. The mRNA and protein levels of JAK2, STAT3, and c-Met were assessed by realtime PCR and western blotting, respectively.
Results. Serum ALT, AST, and TG levels were increased in NASH mice. Histological analysis showed different degrees of steatosis, inflammatory infiltrates, and fibrosis in HFD animals. Exogenous administration of recombinant human (rh) HGF via the tail vein for 14 days markedly decreased ALT and AST to levels lower than those in the control group. Compared with the levels in HFD mice, c-Met, p-c-Met, JAK2, p-JAK2, and p-STAT3 levels were increased in mice that were administered HGF (P < 0.05). Furthermore, silencing of HGF or blocking of its receptor c-Met affected JAK2 and STAT3 protein phosphorylation.
Conclusions. Excess HGF highly probable improved NASH liver function. Combined with its ligand, c-Met, HGF may promote the phosphorylation of JAK2-STAT3 and inhibit inflammation in NASH. Therefore, it may be potentially useful treatment for NASH.
Non-alcoholic fatty liver disease (NAFLD) is a common chronic liver disorder worldwide. Non-alcoholic steatohepatitis (NASH) is a progressive form of NAFLD that is associated with obesity and metabolic syndrome.1 Patients with NASH have the potential to develop liver cirrhosis and hepatocellular carcinoma. However, the mechanism that promotes the progression of NAFLD remains unclear. Hepatocyte growth factor (HGF) is a polypeptide that was originally characterized as a highly potent hepatocyte mitogen. In animal models of various injuries and disease, HGF has been shown to promote cell survival and tissue regeneration and suppress or improve chronic inflammation and fibrosis.2 Mesenchymal-epithelial transition factor (c-Met) functions as the cellular re- ceptor of HGF, and binding of HGF to c-Met induces its dimerization and phosphorylation. Then, intracellular adapter proteins bind to c-Met, which leads to the activation of specific intracellular cascades.3 Previous studies have demonstrated the efficacy of HGF on liver fibrosis/ cirrhosis, and revealed that administration of HGF can prevent the onset of hepatic steatosis in rat liver.4 Tojima5 showed in a NASH mouse model induced by a methionine-choline deficient diet, that HGF ameliorated liver inflammation and fibrosis. Recent studies have reported that the serum HGF levels were significantly higher in NASH mice than in healthy controls, and there is a strong correlation between increased serum fasting HGF levels and the grade and staging of NASH at liver biopsy.6 Additionally, deletion of c-Met in hepatocytes triggers NASH progression.7 However, the detailed molecular mechanism underlying these HGF/c-Met-mediated effects on NASH has not yet been fully elucidated.
Signal transduction and activator of transcription3 (STAT3) is a member of the transcription and activation signal transduction family. Cytokine-induced activation of the Janus-activated kinase2 (JAK2)-STAT3 signalling pathway is critical in inflammatory responses,8 and JAK2-STAT3-mediated apoptosis is involved in hepatocyte protection,9 specifically, phosphorylation STAT3 promotes cell survival and inhibits apoptosis. Although the in vivo protective effects of HGF against liver inflammation and fibrosis caused by exposure to various drugs or chemicals have been reported,4,10 to date, the role of HGF/c-Met in NASH and the relationship between the JAK2-STAT3 signalling pathway and the ameliorative effects of HGF still have not been clarified.
In this study, using RNA interference in a mouse model of NASH, we aimed to test and verify the ameliorative effect of HGF/c-Met and study its relationship with the JAK2-STAT3 signalling pathway.
Material and MethodsAnimals and treatmentSpecific pathogen-free male BALB/c mice (six-weeks old; weight, 18 - 23 g) purchased from Vital River Laboratory Animal Technology (Beijing, China) were randomly assigned to either the control group (n = 6) or the model group (n = 30). Mice in the control group were fed a normal diet (AIN93M, 10% kcal fat), whereas mice in the model group were fed a high-fat diet (HFD, TP23400, 60% kcal fat) for 16 weeks. For the histological analysis, liver samples from each mouse were subjected to hematoxylin and eosin (H&E) staining, and the NAFLD activity score (NAS) was assessed, which includes assessments of hepatic steatosis (0 - 3), inflammation (0 - 3), and hepatocyte ballooning (0 - 2).11 NASH was defined as a NAS score ≥ 5. One experienced pathologist, who was blinded to the purpose of the study, independently interpreted the results. Mice in the model group were randomly assigned to one of five groups. For group B, normal saline was injected via the tail vein daily for the last 14 days of the 16-week modelling; for group C, 0.5 of human recombinant HGF (hrHGF) (R&D Systems, USA) per kilogram of body weight was injected through the tail vein daily for last 14 days; for group D, the c-Met inhibitor PHA665752 (0.15 mg/day) (Pfizer Inc., San Diego, CA)used at the concentrations described and the preliminary experiment,12 was administered via intraperitoneal injection for the last 5 days of the HGF injection regimen; for group E, mice were treated as in group C, and an siRNA against HGF (3.75 nM / 500vl in PBS)13 was injected through the tail vein at 0, 8, 24 h; and for group F, mice were treated as described for group C, and the JAK inhibitor AZD1480 was administered (dose, 30 mg/kg)14 via gavage 20 minutes before HGF injection. Body weights were recorded every week, and the dietary consumption (calorie intake) of each mouse was recorded every day.
At the end of the 16th week, the extent of adiposity and hepatic steatosis in each experimental group was assayed by MRI (PharmaScan 7.0/16 US; Bruker, Germany) After isoflurane (2% v/v) anaesthesia, according to the manufacturer’s protocol, animals were scanned from the diaphragm to the pelvis at 5-mm intervals. After MRI scanning, all mice were fasted for 12 h, and blood was obtained from the tail for blood glucose measurement and insulin-glucose tolerance test (ITT) and oral glucose tolerance test (OGTT) experiments. All animal care and experimentation were performed in accordance with the American Physiological Society’s guiding principles for the care and use of animals, and our study was approved by the ethical committee of Shanxi Medical University.
Serum analysisAll mice were sacrificed, blood was collected, and plasma was immediately recovered and stored at -80 °C for subsequent analyses. Serum levels of alanine aminotransferase (ALT), aspartate aminotransferase (AST), triglyceride (TG), fasting blood-glucose (FBG), and insulin were analysed to assess the extent of liver disease, according to the manufacturers’ instructions. The computational formula for homeostasis model assessment of insulin resistance (HOMA-IR) was HOMA-IR = fasting serum insulin (IU/L) × fasting glucose (mM/L)/22.5. TNF-α, IL-6, and IL-1β contents were measured using ELISA kits obtained from Solarbio (Beijing, China).
siRNA-mediated knockdownTo verify the role of HGF in NASH, four siRNA sequences targeting mouse HGF, a negative control oligo, and a GAPDH control oligo were designed by Shenggong (Shanghai, China) (Table 1). Knockdown efficiency in liver tissue was determined by detecting HGF protein by ELISA.
Sequences of HGF-targeting and control siRNAs.
| Name | Primer | Sequence (5’-3’) |
|---|---|---|
| Hgf-mus-519 | Forward Reverse | CCAUGAAUUUGACCUCUAUTT AUAGAGGUCAAAUUCAUGGTT |
| Hgf-mus-755 | Forward Reverse | GCAAUCCAGAGGUACGCUATT UAGCGUACCUCUGGAUUGCTT |
| Hgf-mus-1147 | Forward Reverse | GGAAUUCCCUGUCAGCGUUTT AACGCUGACAGGGAAUUCCTT |
| Hgf-mus-519 | Forward Reverse | CCUCUUAUUCCUUGGGAUUTT AAUCCCAAGGAAUAAGAGGTT |
| Negative control | Forward Reverse | UUC UCC GAA CGU GUC ACG UTT ACG UGA CAC GUU CGG AGA ATT |
| GAPDH control | Forward Reverse | CAC UCA AGA UUG UCA GCA ATT UUG CUG ACA AUC UUG AGU GAG |
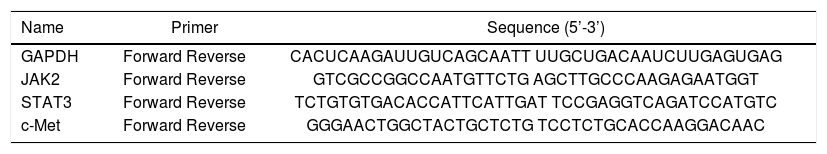
Total RNA was extracted from frozen hepatic tissue, and cDNAs were synthesized using the Fast Quant RT Kit (Tiangen, Beijing, China) from 5 μg of total RNA. Hepatic expression of jak2, Stat3, c-Met, and GAPDH were measured using the SYBR Green Real-Time PCR Kit (Keygenbio, China). The PCR was conducted in a 25 μL volume with the following cycling conditions: 50 °C for 2 min, and 40 cycles of 95 °C for 10 min, 95 °C for 15 s, and 60 °C for 60 s. The final data were used to determine the Ct value. The sequences of the primers are shown in table 2.
Sequences of primers used for quantitative real-time PCR.
| Name | Primer | Sequence (5’-3’) |
|---|---|---|
| GAPDH | Forward Reverse | CACUCAAGAUUGUCAGCAATT UUGCUGACAAUCUUGAGUGAG |
| JAK2 | Forward Reverse | GTCGCCGGCCAATGTTCTG AGCTTGCCCAAGAGAATGGT |
| STAT3 | Forward Reverse | TCTGTGTGACACCATTCATTGAT TCCGAGGTCAGATCCATGTC |
| c-Met | Forward Reverse | GGGAACTGGCTACTGCTCTG TCCTCTGCACCAAGGACAAC |
Tissue lysates containing 40 μg of protein were resolved by SDS-PAGE using 4-20% polyacrylamide gradient gels, and then the fractioned proteins were transferred to nitrocellulose membranes. After blocking with 5% dry milk in a Tris-based saline buffer containing 0.1% Tween 20 for 1 h, membranes were blotted with the corresponding antibodies. The primary antibodies used were: rabbit anti-mouse JAK2/p-JAK2, c-Met/p-c-Met, and STAT3 / p-STAT3 antibodies (Abcam). Band intensity, i.e. the expression of each protein, was measured densitometrically and was normalized to the level of GAPDH.
Statistical analysisThe data are expressed as the mean ± standard deviation (SD). The obtained results were compiled and analysed by Statistical Package for Social Science (SPSS 17.0) using one-way ANOVA followed by Tamhane’s test. Differences with P values < 0.05 were considered statistically significant.
ResultsHFD induces steatohepatitisGross anatomical examination showed that the livers from HFD animals (group B) appeared to be enlarged (hepatomegaly) and paler in colour. Mice in group B also showed significant body weight gain. MRI showed increased subcutaneous fat on the T1 phase and increased visceral fat in the T2 phase in group B mice (Figure 1a).
 induces steatohepatitis in mice. Mice were fed either a HFD or a standard diet (SD) for 16 weeks. a. Gross anatomical features and MRI results. b. Plasma levels of ALT, AST. c. Plasma levels of TG. d. Plasma levels of tumour necrosis factor (TNF)-α, IL-6, and IL-1β. * P < 0.05, ** P < 0.01, and *** P < 0.001 vs. the control group.")
High-fat diet (HFD) induces steatohepatitis in mice. Mice were fed either a HFD or a standard diet (SD) for 16 weeks. a. Gross anatomical features and MRI results. b. Plasma levels of ALT, AST. c. Plasma levels of TG. d. Plasma levels of tumour necrosis factor (TNF)-α, IL-6, and IL-1β. * P < 0.05, ** P < 0.01, and *** P < 0.001 vs. the control group.
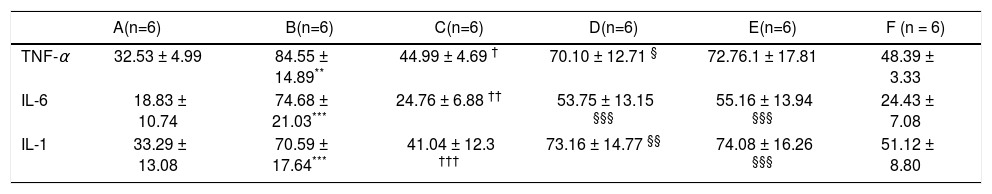
After 16 weeks of HFD feeding, the mice showed liver injury, as reflected by significantly higher transaminase levels compared to those of control mice. The mean ALT, AST, and TG values in group B were 91.68 ± 24.56 U/L, 254.72 ± 29.35 U/L, and 2.11 ± 0.36 mmol/L, respectively, whereas those in the control group were 26.5 ± 0.68 U/L, 88.18 ± 11.26 U/L, and 1.23 ± 0.25 mmol/L, respectively (P1 = 0.019, P2 < 0.001, and P3 = 0.014, respectively). There were statistically significant differences between group B and the control group (Figures 1b and 1c). The average serum levels of TNF-α IL-6, and IL-1β in the control group and group B are shown in table 3, and the levels of these inflammatory cytokines were significantly higher in group B (P1 = 0.003, P2 < 0.001, and P3 < 0.001) (Figure 1d).
Average serum levels of TNF-α, IL - 6, and IL-1 β.
| A(n=6) | B(n=6) | C(n=6) | D(n=6) | E(n=6) | F (n = 6) | |
|---|---|---|---|---|---|---|
| TNF-α | 32.53 ± 4.99 | 84.55 ± 14.89** | 44.99 ± 4.69 † | 70.10 ± 12.71 § | 72.76.1 ± 17.81 | 48.39 ± 3.33 |
| IL-6 | 18.83 ± 10.74 | 74.68 ± 21.03*** | 24.76 ± 6.88 †† | 53.75 ± 13.15 §§§ | 55.16 ± 13.94 §§§ | 24.43 ± 7.08 |
| IL-1 | 33.29 ± 13.08 | 70.59 ± 17.64*** | 41.04 ± 12.3 ††† | 73.16 ± 14.77 §§ | 74.08 ± 16.26 §§§ | 51.12 ± 8.80 |
* P < 0.05
The inhibitory effects of siRNA on HGF protein expression in mouse liver were analysed by ELISA. Based on the data shown in figure 2a, we selected the primers that yielded the lowest average protein content for subsequent tests. Thus, the Hgf-mus-519 siRNAs were used in the RNA interference experiment.
 and serum TNF-α, IL-6, and IL-1 β (d) after siRNA administration in the mice. Group A, the control group; B-F: High-fat diet (HFD) groups (n = 6, each); group B, HFD + normal saline; group C, HDF + rhHDF; group D, HFD + rhHGF + PHA665752 (c-Met inhibitor); group E, HFD + HGF-specific siRNA; and group F, HFD + rhHGF + AZD1480 (JAK inhibitor). * P < 0.05, ** P < 0.01, *** P < 0.001 vs. group A (control);† P < 0.05, †† P < 0.01, ††† P < 0.001 vs. group B (HFD + normal saline); § P < 0.05, §§P < 0.01, §§§ P < 0.001 vs. group C (HFD + HGF).")
a. Determination of HGF protein content in mice injected with different siRNAs by ELISA. The siRNA that yielded the lowest average protein content, Hgf-mus-519, was used in subsequent experiments. Changes in average serum ALT, AST and TG levels (b, c) and serum TNF-α, IL-6, and IL-1 β (d) after siRNA administration in the mice. Group A, the control group; B-F: High-fat diet (HFD) groups (n = 6, each); group B, HFD + normal saline; group C, HDF + rhHDF; group D, HFD + rhHGF + PHA665752 (c-Met inhibitor); group E, HFD + HGF-specific siRNA; and group F, HFD + rhHGF + AZD1480 (JAK inhibitor). * P < 0.05, ** P < 0.01, *** P < 0.001 vs. group A (control);† P < 0.05, †† P < 0.01, ††† P < 0.001 vs. group B (HFD + normal saline); § P < 0.05, §§P < 0.01, §§§ P < 0.001 vs. group C (HFD + HGF).
To better define the impact of the HGF/c-Met system and JAK-STAT3 signalling pathway on NASH, we used a specific HGF RNAi as well as a specific c-Met inhibitor and a JAK2 inhibitor, as mentioned above. Compared with the levels in group C, ALT levels were elevated in groups D and E (P1 = 0.027 and P2 = 0.013), AST levels were elevated in groups D and F (P1 = 0.048 and P2 = 0.002), and TG levels were reduced in groups D and E (P1 = 0.015 and P2 = 0.024) (Figures 2b and 2c).
Serum TNF-α, IL-6, and IL-1 β levels after in vivo RNA interferenceSeveral important cytokines, including TNF-α, IL-6, and IL-1 β, are closely associated with HFD-stimulated hepatic inflammation. The significant up-regulation of these cytokines in the NASH model group and their down-regulation in the various treatment groups were consistent with the different groups as shown in table 3. Compared with the control (group A), TNF-α, IL-6, and IL-1β levels were elevated in group B (P1 = 0.003, P2 < 0.001, P3 < 0.001). Relative to HFD mice (group B), TNF-α, IL- 6, and IL-1 β levels were lower in group C (P = 0.012, P2 = 0.012, P3 < 0.001). Compared with group C (HFD + HGF), TNF- levels were higher in group D (P = 0.05). IL-6 and IL-1 were also higher in groups D and E (P = 0.001, ’P1 P2 < 0.001, < 0.001, P2’ < 0.001) (Figure 2d).
Compared with the control (group A), average hepatic ALT and AST levels in group B were higher, at 91.68 ± 24.56 and 254.72 ± 29.35 U/L, respectively. Hepatic TG levels were increased to 2.11 ± 0.36 mmol/L. In contrast, in group C, the number of lipid droplets decreased, mainly in the perivenular and periportal areas. Consistent with these results, the average hepatic ALT and AST levels in group C decreased to 52.57 ± 5.52 and 70.4 ± 10.27, respectively, in HGF-treated HFD mice. Therefore, HGF administration improved NASH during HFD treatment.
Histological analysis showed that compared to the control mice in group A, mice in group B were full of fat droplets and inflammatory cells (Figure 3). In group C mice, there were only slight signs of fatty liver disease, whereas mice in groups D, E, and F displayed massive fatty liver degeneration.
 after treatment. More severe steatohepatitis was observed in HFD mice when HGF, c-Met, or JAK2 was inhibited. A. Control group. B-F. Different intervention groups as shown in figure. * P < 0.05, ** P < 0.01, *** P < 0.001 vs. group A (control);† P < 0.05, †† P < 0.01, ††† P < 0.001 vs. group B (HFD + normal saline); § P < 0.05, §§ P < 0.01, §§§ P < 0.001 vs. group C (HFD + HGF).")
Liver H&E staining (200x) after treatment. More severe steatohepatitis was observed in HFD mice when HGF, c-Met, or JAK2 was inhibited. A. Control group. B-F. Different intervention groups as shown in figure. * P < 0.05, ** P < 0.01, *** P < 0.001 vs. group A (control);† P < 0.05, †† P < 0.01, ††† P < 0.001 vs. group B (HFD + normal saline); § P < 0.05, §§ P < 0.01, §§§ P < 0.001 vs. group C (HFD + HGF).
As shown in figure 4A, mice in group C showed significant c-Met, jak2, and stat3 up-regulation compared to the corresponding levels in group B mice. Blockage of the HGF receptor or silencing of HGF decreased jak2 and stat3 expression, and the expression of jak2 and stat3 in groups D and E was significantly lower than that in group C; however, c-Met RNA levels were up-regulated.
;† P < 0.05,†† P < 0.01, ††† P < 0.001 vs. group B (HFD + normal saline); § P < 0.05, §§ P < 0.01, §§§ P < 0.001 vs. group C (HFD + HGF).")
RNA, protein andphosphorylatedprotein expression of c-Met, JAK2, STAT3 in various treatment groups. a. After treatment, mRNA levels of c-Met, JAK2, and STAT3 in the livers of NASH mice at the end of treatment were compared. GAPDH Ct value 17-18. b. Liver lysates were immunoblotted for phosphorylated and total c-Met, JAK2, and STAT3, GAPDH was used as a loading control. Representative results from at least three experiments are shown. c. Average c-Met, JAK, and STAT3protein expression. d. Averagep-c Met, p-JAK, andp-STAT3 expression. *P < 0.05, **P < 0.01, ***P < 0.001 vs.group A (control);† P < 0.05,†† P < 0.01, ††† P < 0.001 vs. group B (HFD + normal saline); § P < 0.05, §§ P < 0.01, §§§ P < 0.001 vs. group C (HFD + HGF).
Western blotting showed that exogenous administration of rhHGF can stimulate the expression of c-Met, JAK2, and STAT3 (P1 = 0.024, P2 and P3 < 0.001) in NASH tissue (Figure 4b). In mice that were administered PHA665752 or HGF-siRNA, the levels of c-Met and JAK2 protein were down-regulated. Furthermore, STAT3 expression was decreased in groups D, E, and F (Figure 4c). To further explore the mechanism underlying these findings, we examined the expression of the phosphorylated-STAT3, c-Met, and JAK2 in NASH mice with or without AZD1480 treatment. The results showed that AZD1480 decreased p-STAT3 and p-JAK2 levels in the liver of NASH mice, but increased p-c-Met levels (Figure 4d).
DiscussionNASH is a major public health challenge in developed countries. However, the multifactorial mechanisms governing the progression from the initial phase of simple fat accumulation to the later stages of the disease are still poorly defined. Our experiments indicated that the model group exhibited metabolic disorders and liver pathology characteristic of steatohepatitis with increased ALT and AST activity after 16 weeks of HFD feeding.
As a multifunctional molecule, HGF binding to its cell surface receptor c-Met, can induce phosphorylation of c-Met at several tyrosine residues, which in turn provides docking sites for several intracellular signalling mole-cules.15 Our results showed that excess HGF can alleviate liver lesions and promote the recovery of liver function in NASH mice. Both the serological and pathological alterations confirmed that HGF administration could be a promising solution for NASH. Thus, the HGF/c-Met system has direct implications in the pathogenesis of NASH, and it appears to be hepatoprotective.
It was previously reported that a continuous infusion of rhHGF relieves the hepatitis and liver fibrosis in methionine- and choline-deficient NASH model mice.5 Other reports showed that HGF prevented the onset of hepatic steatosis in rat liver.4 These results, which demonstrate the ability of rhHGF to alleviate the pathological changes in NASH livers and contribute to the recovery of liver function, suggest its possible efficacy for the treatment of NASH. Furthermore, Minoru16 showed that the HGF/c-Met system up-regulates apolipoprotein apoB synthesis, promotes fat mobilization, and moves a lot of fat from the liver to the blood, resulting in increased serum TG levels. Serum TG was also increased in our experiment model; thus, our results verify the conclusions of these previous studies.
Our results also showed that:
- •
c-Met and p-c-Met protein levels were increased, and p-JAK2 and p-STAT3 levels were reduced in NASH mice.
- •
c-Met, JAK2, and STAT3 mRNA and protein levels were diminished after knockdown of HGF in vivo.
These results implied that the HGF/c-Met system is activated, but the JAK2-STAT3 signalling pathway is restrained in NASH mice. NAFLD patients commonly show leptin resistance,17 which promotes lipid synthesis in the liver, leading to the development of fatty liver. The underlying reason may be that leptin secretion declines during NASH, which suppresses the JAK2-STAT3 signalling pathway and increases fat synthesis, thus aggravating inflammation.
We investigated the mechanisms underlying the ameliorating effects of HGF on NASH, and our results showed that rhHGF stimulated the recovery of liver function and improved the pathological changes. Blood analyses indicated that interference of HGF expression using siRNA in NASH mice is deleterious for the recovery of liver functions.
RNA interference experiments revealed that injection of HGF-specific siRNA inhibited the signalling downstream of the HGF receptor (c-Met), and reduced the expression of c-met, jak2, and stat3. IL-6, the key cytokine stimulus of JAK2-STAT3, is over-expressed in NASH model mice, and IL-6 levels in groups B, D, and E were increased compared to the control. Compared to group B, IL-6 levels in group F mice were decreased. A study of tumours showed that IL-6 interacts with IL-6R/gp130, resulting in JAK/STAT3 activation, which in turn increases the expression of autocrine IL-6.18 It is interesting to note that the control group not only had lower transaminase levels but also lower IL-6 levels and higher Stat3 expression levels. These observations suggest a context- or microenvironmentdependent role for activation of the IL-6-JAK-STAT3 signalling pathway in the regulation of NASH.
Leptin is the main regulator of fat stores. Through its interaction with JAK2-STAT3, it causes various biological effects, such as decreasing TG levels, elevating the insulin sensitivity of the liver and peripheral tissues, and reducing fat deposition.19 In our study, administration of HGF in NASH mice increased TG levels, whereas inhibition of HGF decreased TG levels. This suggests that the ameliorating effect of rhHGF on NASH was related to the JAKSTAT3 signalling pathway.
STAT3 is widely expressed in different types of cells and tissues, where it participates in various physiological functions, such as cell growth, transformation, and apoptosis regulation. JAK2-STAT3 is common pathway for the transduction of numerous inflammatory factor signals, and phosphorylated JAK2 is the main factor promoting STAT3 phosphorylation. Activation of gp130-STAT3 in the development of NAFLD can be effective against fat stress.20 JAK2 and STAT3 were significantly expressed in NASH mice, as shown by RT-PCR, and these data were confirmed at the protein level. The specificity of these effects for the JAK2-STAT3 pathway was confirmed by the administration of the JAK2 blocker AZD1480. This implies that HGF/c-Met signalling, which is upstream of the JAK2-STAT3 pathway, has a corresponding biological effect through other signalling pathways. This result is consistent with the findings of Toshikazun.21 Furthermore, administration of AZD1480 induced a decrease in JAK2 protein and phosphorylation levels, leading to a decrease in STAT3 phosphorylation levels and inhibition of the JAK2-STAT3 pathway signalling and inhibition of corresponding biological functions. Our results indicate that administration of exogenous HGF can increase the levels of its specific ligand c-Met, activate the HGF/c-Met system, promote phosphorylation of JAK2-STAT3 signalling pathway components, and increase gene and protein levels. Blocking the HGF receptor c-Met or silencing HGF can decrease c-Met, JAK2, and STAT3 gene and protein levels. This suggests that the JAK2-STAT3 pathway plays a critical role in the ameliorating effect of rhHGF on NASH. These results confirm that over-expression of HGF, combined with its ligand c-Met, can promote the phosphorylation of JAK2-STAT3, and through JAK2-STAT3 phosphorylation and signalling, NASH inflammation was effectively inhibited.
It is important to note that HGF/c-Met signalling is crucial for normal embryonic development and adult tissue repair in mammals, and is maintained at a certain level in human normal tissue.22 However, improper amplification of the c-Met gene and activation of HGF/c-Met signalling, along with elevated protein expression and constitutive kinase activation, may result in growth, invasion, migration, and tumourigenesis.23
Another explanation for the anti-inflammatory effect of exogenous HGF is probable associated with IL-10. IL-10 -/mice displayed greater liver inflammatory response but less steatosis after HFD feeding, and inflammation-associated IL-6 / STAT3 activation contributes to the reduced steatosis.24 In addition, the systemic levels of TNF-α increased, while IL-10 decreased in accordance with the severity of NAFLD, which supports a role for systemic inflammatory mediators in promoting steatosis progression.25 The potential mechanisms involved in the antihepatitis properties of HGF have been identified. Reciprocal regulation of cytokine profiles by HGF, that is the suppression of proinflammatory cytokines and induction of antiinflammatory cytokines, has also been reported in animal models of endotoxemia,26 cardiac allograft transplantation27. In those studies, HGF suppressed proinflammatory cytokines, including MCP-1, IL-6, IL-1β, and interferon-γ, and it induced anti-inflammatory IL-10. More importantly, increasing HGF expression upregulates IL-10 in liver injury.28 It could be excess HGF upregulate IL-10, which played the anti-inflammatory effects in NASH models. Of course, this requires further studies.
In conclusion, HGF is an effective agent for relieving HFD-induced NASH. High levels of HGF alleviate the predisposing factors of liver steatosis, and consequently protect the host from NASH. These effects were accompanied by activation of the JAK2-STAT3 signalling pathway and inhibition of the inflammatory response in NASH. The results of this study suggest clinical trials aimed at targeting HGF in patients with NASH. However, further studies are required to elucidate the precise mechanism underlying the inhibitory effect of HGF on NASH.
Abbreviations- •
ALT: alanine aminotransferase.
- •
AST: aspartate transaminase.
- •
HGF: hepatocyte growth factor.
- •
NASH: non-alcoholic steatohepatitis
- •
c-Met: mesenchymal-epithelial transition factor.
- •
FBG: fasting blood-glucose.
- •
HFD: high-fat diet.
- •
JAK2: janus-activated kinase2.
- •
NAFLD: Non-Alcoholic Fatty Liver Disease.
- •
STAT3: signal transduction and activator of transcription3.
- •
TG: triglyceride.
This work was supported by the Health and Family Planning Commission Foundation of Shanxi Province, China (No.2015029). Science and Technology Department Foundation of Shanxi Province, China (No. 201601D021148). Provincial scientific instruments sharing funds, jin cai teach (2016) 76. Shanxi medical university of science and technology innovation fund projects in 2011, and the school word (2012) 11. Scientific and technological achievements transformation guidance special of Shanxi Province, China (No. 201604D132042).
AcknowledgmentsWe thank the Department of Otorhinolaryngology Head and Neck Tumor key Laboratory of Shanxi Province, Taiyuan, Shanxi, China.
Disclosure of Conflict of InterestNone.



