




Caffeine consumption is associated with beneficial effects on hepatic disorders. The objectives of this study were to evaluate the antifibrotic effects of caffeine on experimental nonalcoholic steatohepatitis (NASH) induced with a high-fat, high-sucrose, high-cholesterol diet (HFSCD), as well as to evaluate the ability of caffeine to prevent the progression of experimental liver fibrosis induced by the administration of thioacetamide (TAA) in rats and explore the mechanisms of action.
MethodsNASH and fibrosis were induced in rats by the administration of an HFSCD for 15 weeks, and liver fibrosis was induced by intraperitoneal administration of 200 mg/kg TAA 3 times per week, for 6 weeks. Caffeine was administered at a dose of 50 mg/kg body weight. The effects of diet, TAA, and caffeine on fibrosis were evaluated by biochemical and histological examinations. The profibrotic pathways were analyzed by western blotting and immunohistochemistry.
ResultsRats exhibited liver fibrosis after HFSCD feeding and the administration of TAA. Caffeine could reduce the hepatic level of collagen and the fibrotic area in the liver. Caffeine prevented the progression of liver fibrosis by decreasing transforming growth factor-beta (TGF-β), connective tissue growth factor (CTGF), and alpha-smooth muscle actin (α-SMA) expression and by inhibiting the activation of mitogen-activated protein kinases (MAPKs) and Smad3 phosphorylation.
ConclusionsCaffeine attenuates NASH and the progression of liver fibrosis due to its antifibrotic effects and modulating the MAPK and TGF-β pathways. Therefore, caffeine could be a suitable candidate for treating liver diseases associated with fibrosis.
Because of the role that the liver plays in xenobiotic detoxification, this organ is continuously exposed to factors that may induce damage [1]. Repetitive tissue damage and chronic inflammation result in events that can trigger liver fibrosis. Fibrogenesis is a dynamic process related to the degree of liver parenchymal damage, involves different cellular and molecular disorders and is associated with alterations in the amount and composition of the extracellular matrix (ECM) [2–4]. Among the main causes of liver fibrosis, alcoholic liver disease is prominent, and in recent decades, nonalcoholic steatohepatitis (NASH) has become very important [5–7]. On the other hand, liver functions can be compromised by an increase in the production rate of reactive oxygen species, which may be associated with the elevated consumption of alcohol or the occurrence of NASH [8]. Regardless, hepatocytes are targets of hepatotoxic agents [2,9]. Injured and apoptotic hepatocytes produce chemoattractant signals for immune cells that produce inflammatory and profibrogenic cytokines such connective tissue growth factor (CTGF) and transforming growth factor-beta (TGF-β) [2,10]. TGF-β is the most potent profibrogenic cytokine and a key driver of hepatic stellate cell (HSC) activation and liver fibrosis [11]. The profibrogenic effects of TGF-β are mediated by pathways that depend on the transcription factor R-Smad and by mitogen-activated protein kinase (MAPK) [11,12]. Upon activation, HSCs express myogenic markers such as alpha-smooth muscle actin (α-SMA) and produce large amounts of ECM proteins, leading to fibrosis [11]. Alcoholic liver disease and NASH are common liver diseases that may progress to fibrosis, cirrhosis, and hepatocellular carcinoma (HCC); however, no effective therapy is currently available for these pathologies [5-7,13]. Therefore, there is an urgent need to discover therapeutic targets and develop effective therapies to prevent, attenuate, slow or reverse alcohol- or NASH-induced fibrosis.
Several herbal medicines have been reported to have important beneficial effects on liver diseases [14]. One of the best of these compounds is caffeine, whose liver benefits and mechanisms have been studied by us and other researchers [15]. The hepatoprotective properties of caffeine have been observed in different models of experimental liver damage, such as bile duct ligation and carbon tetrachloride (CCl4) administration, in which this alkaloid exerted important antioxidant, anti-inflammatory, anticholestatic, and antifibrotic effects [16–19]. Caffeine consumption in humans and animals has been reported to be associated with a reduction in liver damage, and regular consumption of caffeine in coffee can significantly attenuate liver fibrosis in patients with hepatic steatosis [20–26]. Therefore, we hypothesized that caffeine prevents the development of liver fibrosis by attenuating the profibrogenic TGF-β and MAPK signaling pathways in an experimental model of NASH and TAA-induced liver damage. Consequently, this study aimed to investigate the potential hepatoprotective effect of caffeine in an experimental NASH model and to elucidate the molecular mechanisms involved in this protection, as well as to evaluate the capacity of caffeine to slow the progression of liver fibrosis in a thioacetamide (TAA)-induced model of chronic liver damage.
2Materials and methods2.1ReagentsCaffeine, cholesterol, cholate, and TAA were purchased from Sigma-Aldrich® (Missouri, USA). Gloria® unsalted butter was used as a source of sucrose. Zulka® icing sugar was used, and the casein used was the Rennet Casein Irish Dairy Board brand.
2.2Animal treatments2.2.1Nonalcoholic steatohepatitisNASH was induced in male Wistar rats with initial body weights of 100-120 g with a high-fat, high-sucrose, high-cholesterol diet (HFSCD) as previously reported [27], ingredients are shown in Table 1. The rats were randomly divided into four groups of 8 rats each. The control group was fed a control diet (Labdiet® No. 5053, Indiana, USA) ad libitum for 15 weeks; the HFSCD group was fed the HFSCD ad libitum for 15 weeks; the HFSCD + caffeine group received HFSCD ad libitum plus 50 mg of caffeine/kg body weight daily by gavage; and the caffeine-only group received 50 mg of caffeine/kg body weight daily by gavage. Bodyweight gain was assessed once per week. The animals had free access to water and were housed in polycarbonate cages under controlled conditions (21 ± 1°C, 50%‐60% relative humidity and 12‐hour dark/light cycles). Body weights were assessed at the end of the treatments. After 15 weeks of treatment, the rats were anesthetized with ketamine and xylazine, and then, blood was collected by cardiac puncture and centrifuged in tubes at 3000 rpm (12,000 g). The liver was rapidly removed and stored at -75°C.
High-fat, high-sucrose, high-cholesterol diet composition.
To evaluate the effect of caffeine on the progression of liver fibrosis, male Wistar rats (100-120 g) were randomly divided into five groups of 8 rats each. The control group received 1 mL of tap water daily intraperitoneally (TAA vehicle). The TAA-3-Week and TAA-6-Week groups were intraperitoneally administered 200 mg/kg TAA 3 times per week for 3 and 6 weeks, respectively, as described previously [28]. The TAA-6-Week + caffeine group were administered TAA for three weeks, after which 50 mg/kg caffeine was administered by gavage daily for three weeks. The caffeine-only group received 50 mg of caffeine/kg of body weight daily by gavage. Animals had free access to food (Labdiet® No. 5053, Indiana, USA) and water, were housed in polycarbonate cages under controlled conditions (21 ± 1°C, 50%‐60% relative humidity and 12‐hour dark/light cycles). Liver and body weight were assessed at the end of the treatments. After six weeks of treatment, the rats were anesthetized with ketamine and xylazine, and then, blood was collected by cardiac puncture and centrifuged in tubes at 3000 rpm (12,000 g). The liver was rapidly removed, weighed, and stored at -75°C.
The study complies with the institution's guidelines and the Mexican official regulations (NOM-062-ZOO-1999) regarding the technical specifications for the production, care, and handling of laboratory animals. The approval number provided by the Cinvestav Ethics Committee is 281-19.
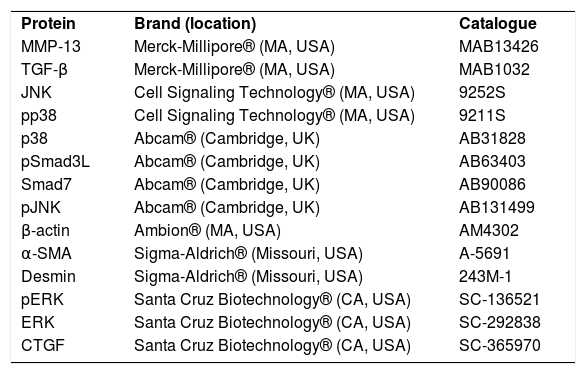
2.3AntibodiesThe antibodies used were pSmad3, Smad7, c-Jun N-terminal kinases (JNK), extracellular signal-regulated kinases (ERK), p38, pJNK, pERK, pp38, metalloproteinase (MMP) 13, TGF-β, α-SMA, β-actin, desmin and CTGF and are described in Table 2. The dilutions used were as follows: western blotting (1:500) and IHC (1:250).
Antibodies used in western blot and immunohistochemistry techniques.
Collagen concentrations were determined by measuring the hydroxyproline levels in fresh liver samples after the samples were digested with dimethylaminobenzaldehyde in an acid solution (Ehrlich's reagent), and the absorbance was measured at 560 nm [29] as described previously [30].
2.5Histological examinationsLiver tissues were fixed in 4% paraformaldehyde in phosphate-buffered saline (PBS). Then, the tissue samples were embedded in paraffin, and 5-μm-thick sections were prepared. The sections were prepared for immunohistochemistry or stained with Masson´s trichrome staining for histological analysis. Liver slices from 3-4 rats of each group were quantified; then, three different fields were analyzed with a 10x objective, blue staining was quantified. All stained slides were visualized using a light microscope (80i, Eclipse, Nikon®, Tokyo, Japan).
2.6Immunohistochemical assaysImmunohistochemical (IHC) staining was performed using an immunoperoxidase protocol. Then, the samples were incubated with a primary antibody diluted in 3% fetal bovine serum overnight. Next, the samples were rinsed with 1× PBS) five times for five minutes each. The antibodies used for IHC were against TGF-β and α-SMA and are described in Table 2. The stained specimens were covered with resin and allowed to dry for two days. All stained slides were visualized using a light microscope (80i, Eclipse, Nikon®, Tokyo, Japan). Liver slices from 3-4 rats of each group were quantified; then, three different fields were analyzed with a 10x objective, brown staining was considered a positive reaction. Digital images of the histological sections were taken, and positive signals were quantified with ImageJ® software (NIH, MD, USA) [31].
2.7Western blottingFor protein analysis by western blotting, liver tissue was homogenized in lysis buffer (Sigma-Aldrich®, Missouri, USA) supplemented with protease and phosphatase inhibitor cocktails (Sigma-Aldrich®, Missouri, USA) and then centrifuged at 12,000 rpm (13,200 g) for twenty minutes at 4°C. The supernatant was collected, and the protein concentration was measured using the bicinchoninic acid method [32] (Pierce BCA Protein Assay, catalog number 23223, Thermo Fischer Scientific®, NY, USA). Equal amounts of protein (40 μg) were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis using a 12% gel (100 V, 3 h, room temperature), and the proteins were electrotransferred onto a 0.45 µm immuno-Blot PVDF membrane (BIO-RAD®, CA, USA) (0.25 A, 1.40 h at 4°C). The membranes were blocked using 5% bovine serum albumin (Sigma-Aldrich®, Missouri, USA) in tris-buffered saline with Tween-20 for two hours at room temperature. The membranes were incubated in a 500-fold diluted solution of primary antibodies overnight at 4°C and then incubated in a 5000-fold diluted solution of secondary antibodies for two hours at room temperature. The primary antibodies against pSmad3, Smad7, JNK, ERK, p38, pJNK, pERK, pp38, MMP 13, TGF-β, α-SMA, β-actin, desmin, and CTGF are described in Table 2. An antibody against β-actin was used as the internal control, and the results are expressed as a ratio compared to the control. The membranes were bathed in luminol reagent (Santa Cruz Biotechnology®, CA, USA) for development. Photographic plates (catalog number 822526, Kodak®, NY, USA) were digitized, and the intensity of each band was quantified using densitometric scanning with ImageJ® software (NIH, MD, USA) [33].
2.8ZymographyProteolytic activity was evaluated on gelatin-substrate gels. Hepatic tissue (0.25 g) was homogenized, sonicated, and centrifuged with 1.7 mL of 1× PBS at 13,000 rpm (16,400 g) for ten minutes. The supernatant was collected, and the proteins were quantified by the bicinchoninic acid method [32]. Equivalent volumes were added to 50 μg of protein in sample buffer without a reducing agent and injected into eight percent acrylamide gels polymerized with 1 mg/mL gelatin.
Electrophoresis was then performed at 72 V for two hours. Proteolytic activity was detected as transparent bands against the background staining of the undigested substrate in the gel at the expected position according to the molecular weight of the MMPs. The images were digitized, and the intensity of each band was quantified with ImageJ® software (NIH, MD, USA) [33].
2.9Statistical analysesAll graphical data are shown as the mean ± standard error (SE). Multiple comparisons were performed using GraphPad Prism® 7.0 software (CA, USA). The results were analyzed using one-way ANOVA to compare multiple groups, followed by Tukey's test. P≤ 0.05 was considered statistically significant.
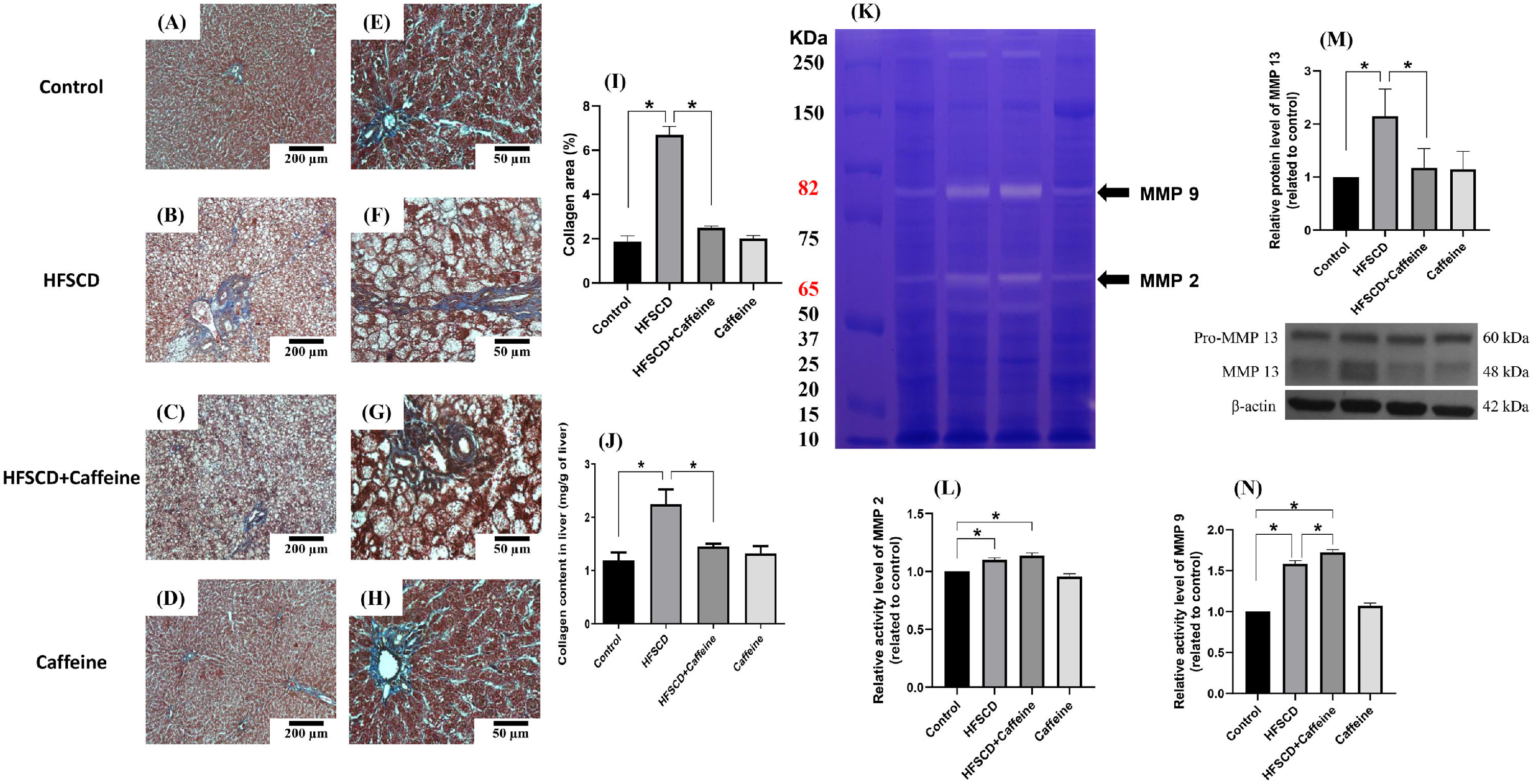
3Results3.1Caffeine prevents the development of liver fibrosis induced by an HFSCDECM deposition was visualized by Masson´s trichrome staining of liver samples and estimated by measuring the hydroxyproline level (Fig. 1). HFSCD feeding produced a significant increase in the level of collagen, and caffeine consumption prevented this effect (Fig. 1I and J). ECM degradation was determined by measuring the activities of MMP 2 and MMP 9 and the protein levels of MMP 13 (Fig. 1K, L, N). The activities of both gelatinases (MMP 2 and 9) were significantly increased in HFSCD-fed rats compared to control rats, and caffeine significantly increased the activity of MMP 9 in HFSCD-fed rats but did not increase the activity of MMP 2. The MMP 13 protein levels were estimated by western blotting with specific antibodies and were increased in the HFSCD group, while caffeine consumption prevented this elevation (Fig. 1M). Caffeine alone induced no effects.
. Effect of caffeine as shown by Masson's trichrome staining in the livers; Control (A)(E), HFSCD (B)(F), HFSCD + Caffeine (C)(G) and Caffeine (D)(H). Scale bars = 200 μm and 50 μm. The percentages of the collagen areas in histological sections are shown (I) (n = 4). Collagen levels were determined by measuring the liver hydroxyproline level (J) (n = 6). The activities of MMP 2 (L) and MMP 9 (N) were determined by zymography (K) (n = 5). The values are presented as fold increases in optical density values normalized to the values of the control group (control = 1). Protein levels of MMP 13 (M) in liver tissue samples were examined by western blot analysis (n = 3), and β-actin was used as a control. Each bar represents the mean value ± SE. (*) indicates P < 0.05.")
Caffeine prevents fibrosis and profibrogenic mediators and regulates extracellular MMPs in rats fed a high-fat, high-sucrose, high-cholesterol diet (HFSCD). Effect of caffeine as shown by Masson's trichrome staining in the livers; Control (A)(E), HFSCD (B)(F), HFSCD + Caffeine (C)(G) and Caffeine (D)(H). Scale bars = 200 μm and 50 μm. The percentages of the collagen areas in histological sections are shown (I) (n = 4). Collagen levels were determined by measuring the liver hydroxyproline level (J) (n = 6). The activities of MMP 2 (L) and MMP 9 (N) were determined by zymography (K) (n = 5). The values are presented as fold increases in optical density values normalized to the values of the control group (control = 1). Protein levels of MMP 13 (M) in liver tissue samples were examined by western blot analysis (n = 3), and β-actin was used as a control. Each bar represents the mean value ± SE. (*) indicates P < 0.05.
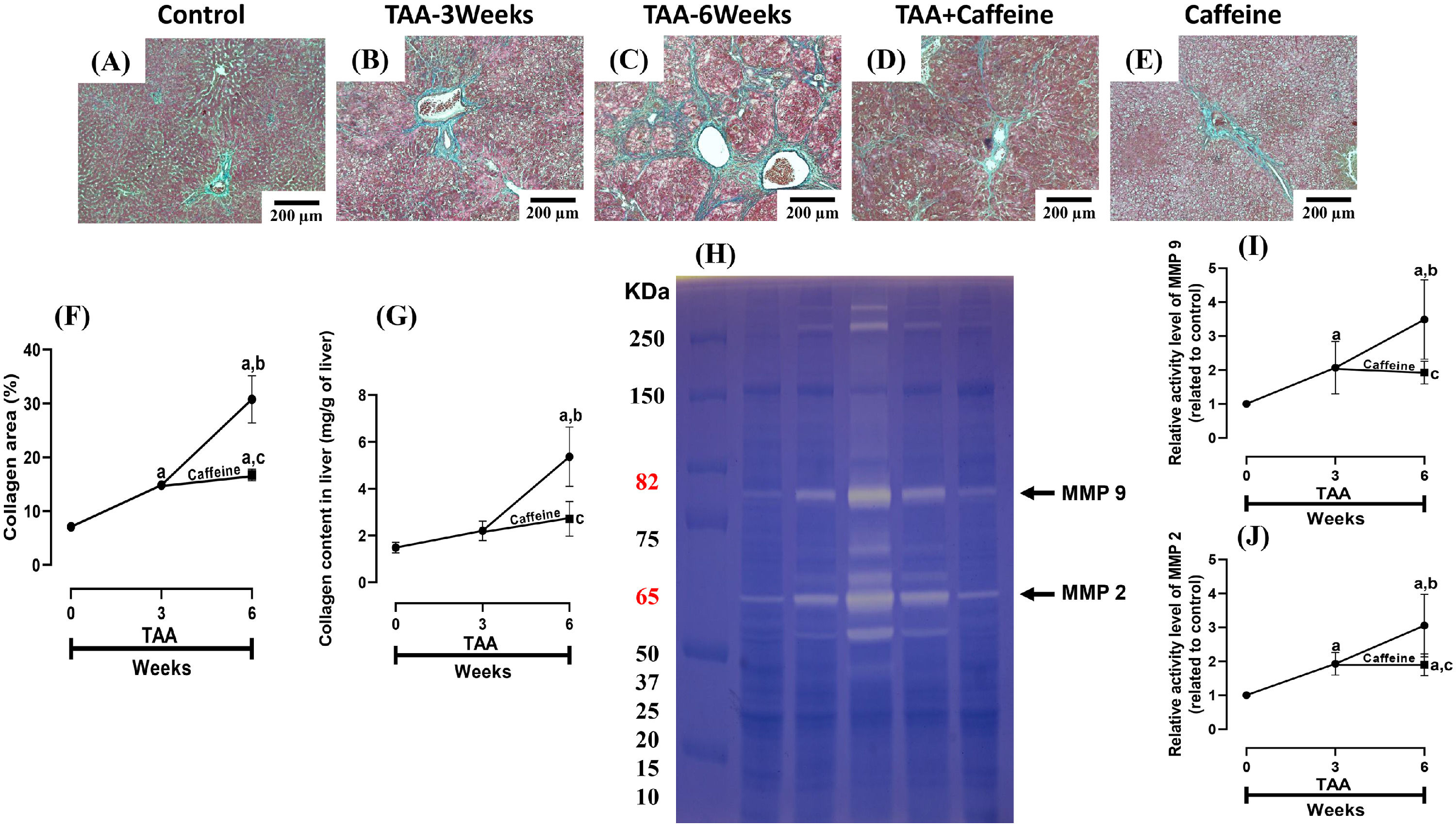
ECM accumulation in liver samples was evaluated by Masson´s trichrome staining and estimated by measuring the hepatic hydroxyproline level (Fig. 2). As shown in Figure 2 (F, G), chronic treatment with TAA increased the hepatic collagen level, and caffeine administration from Weeks 3 to 6 attenuated the effect of TAA. ECM degradation was determined by measuring the activities of MMP 2 and 9 (Fig. 2H-J), which were increased in TAA-6-Week group rats, and caffeine consumption attenuated these effects. Caffeine alone induced no effects.
, TAA-3-week-treated (B), TAA-6-week-treated (C), TAA + caffeine-treated (D), and caffeine-treated (E) rats. Scale bar = 200 μm. The percentages of the collagen areas in histological sections are shown (F) (n = 3). Collagen levels were determined by measuring the liver hydroxyproline level (G) (n = 6). The activities of MMP 2 (J) and MMP 9 (I) was determined by zymography (H) (n = 7). The values are presented as fold increases in optical density values normalized to the values of the control group (control = 1). Each bar represents the mean value ± SE. (a) P < 0.05 compared with the control group; (b) P < 0.05 compared with the TAA-3-Week group; (c) P < 0.05 compared with the TAA-6-Week group.")
Caffeine abrogates fibrosis and profibrogenic mediators and regulates extracellular MMPs in rats with TAA-induced cirrhosis. Effect of caffeine as shown by Masson's trichrome staining in the livers of control (A), TAA-3-week-treated (B), TAA-6-week-treated (C), TAA + caffeine-treated (D), and caffeine-treated (E) rats. Scale bar = 200 μm. The percentages of the collagen areas in histological sections are shown (F) (n = 3). Collagen levels were determined by measuring the liver hydroxyproline level (G) (n = 6). The activities of MMP 2 (J) and MMP 9 (I) was determined by zymography (H) (n = 7). The values are presented as fold increases in optical density values normalized to the values of the control group (control = 1). Each bar represents the mean value ± SE. (a) P < 0.05 compared with the control group; (b) P < 0.05 compared with the TAA-3-Week group; (c) P < 0.05 compared with the TAA-6-Week group.
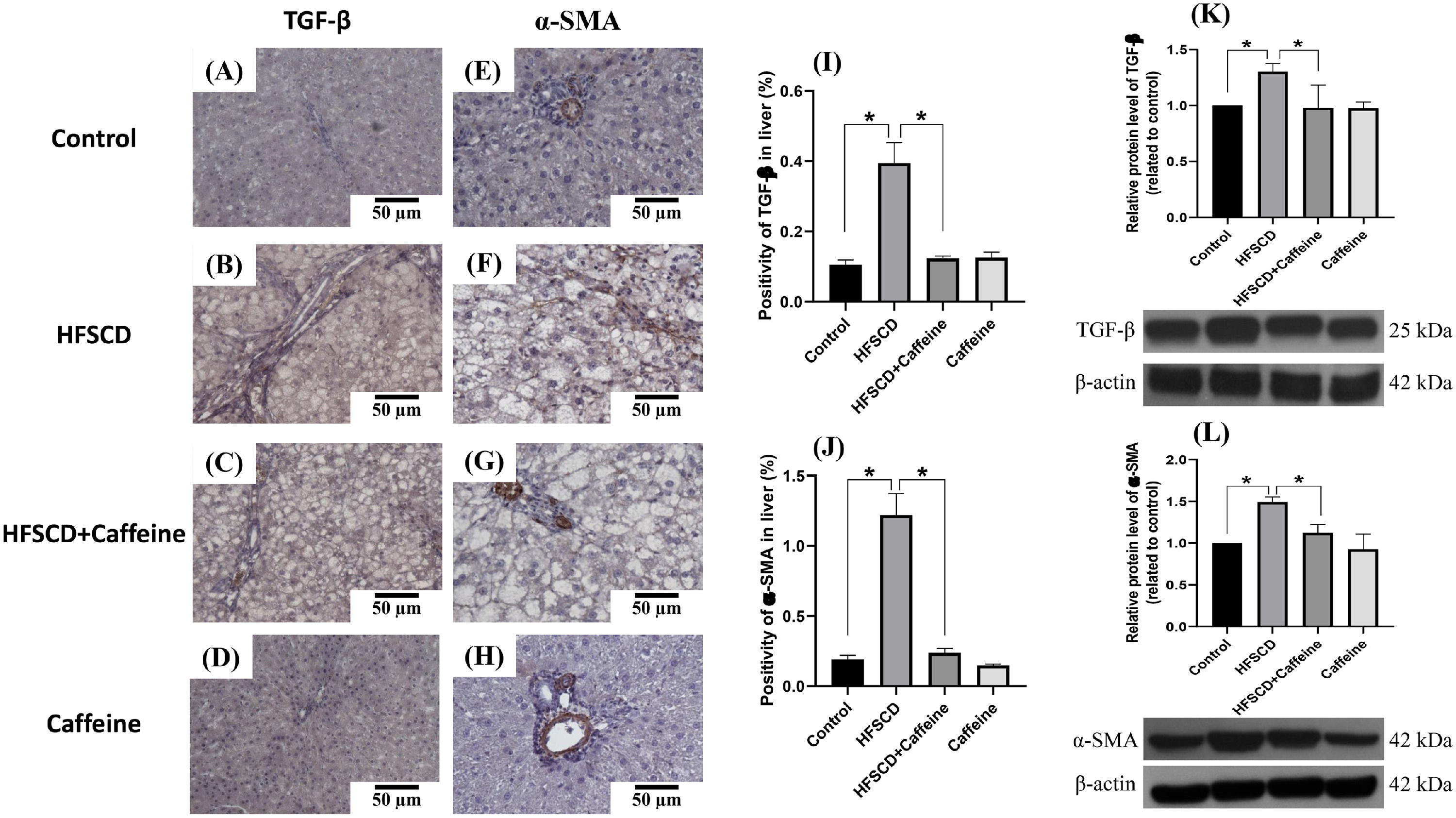
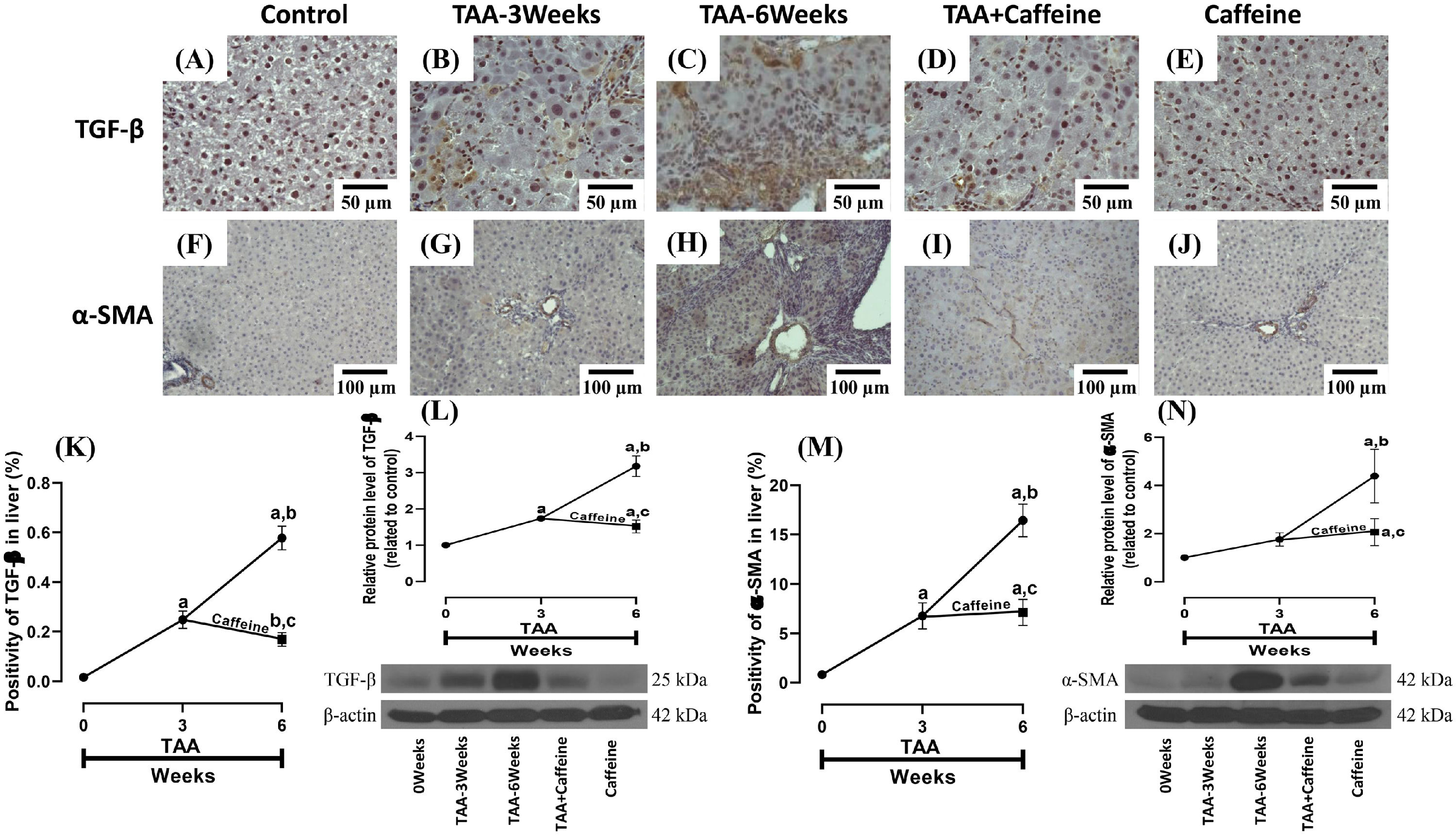
Figure 3 shows the immunohistochemical and western blot analysis of TGF-β and α-SMA in HFSCD-fed rats. The HFSCD group had significantly increased levels of both proteins (Fig. 3I-L). Notably, caffeine consumption completely prevented the increases in these profibrogenic proteins in HFSCD-fed animals (Fig. 3I-L). Figure 4 shows the immunohistochemical and western blot analysis of TGF-β and α-SMA in TAA-treated rats. The TAA-6-Week group had significantly increased protein levels of both factors relative to those of the control group (Fig. 4K-N). Importantly, caffeine consumption significantly attenuated the increases in these proteins in comparison with those in the TAA-6-Week group (Fig. 4K-N). Caffeine alone induced no effects.
. Representative immunohistochemical images of TGF-β in the Control (A), HFSCD (B), HFSCD + Caffeine (C) and Caffeine groups (D) and α-SMA in the Control (E), HFSCD (F), HFSCD + Caffeine (G) and Caffeine groups (H) are shown. Scale bar = 50 μm. The positive area of TGF-β is shown in the histogram (I), and the positive area of α-SMA is shown in the histogram (J) (n = 4). The protein levels of TGF-β (K) and α-SMA (L) in liver tissue samples were determined by western blot analysis (n = 3), and β-actin was used as a control. The values are presented as fold increases in optical density values normalized to the values of the control group (control = 1). Each bar represents the mean value ± SE. (*) indicates P < 0.05.")
Caffeine maintains the basal levels of TGF-β and α-SMA in rats fed a high-fat, high-sucrose, high-cholesterol diet (HFSCD). Representative immunohistochemical images of TGF-β in the Control (A), HFSCD (B), HFSCD + Caffeine (C) and Caffeine groups (D) and α-SMA in the Control (E), HFSCD (F), HFSCD + Caffeine (G) and Caffeine groups (H) are shown. Scale bar = 50 μm. The positive area of TGF-β is shown in the histogram (I), and the positive area of α-SMA is shown in the histogram (J) (n = 4). The protein levels of TGF-β (K) and α-SMA (L) in liver tissue samples were determined by western blot analysis (n = 3), and β-actin was used as a control. The values are presented as fold increases in optical density values normalized to the values of the control group (control = 1). Each bar represents the mean value ± SE. (*) indicates P < 0.05.
, TAA-3-week-treated (B), TAA-6-week-treated (C), TAA + caffeine-treated (D), and caffeine-treated (E) rats are shown; scale bar = 50 μm. Representative immunohistochemical images of α-SMA in Control (F), TAA-3-Week-treated (G), TAA-6-Week-treated (H), TAA + Caffeine-treated (I), and Caffeine-treated (J) rats are shown, scale bar = 100 μm. The positive area of TGF-β is shown (K) (n = 3). The protein level of TGF-β in liver tissue samples was determined by western blot analysis (L) (n = 3), and β-actin was used as a control. The positive area of α-SMA is shown (M) (n = 3). The protein level of α-SMA in liver tissue samples was determined by western blot analysis (N) (n = 3), and β-actin was used as a control. The values are presented as fold increases in optical density values normalized to the values of the control group (control = 1). Each bar represents the mean value ± SE. (a) P < 0.05 compared with the control group; (b) P < 0.05 compared with the TAA-3-Week group; (c) P < 0.05 compared with the TAA-6-Week group.")
Caffeine maintains the basal levels of TGF-β and α-SMA in rats with TAA-induced cirrhosis. Representative immunohistochemical images of TGF-β in control (A), TAA-3-week-treated (B), TAA-6-week-treated (C), TAA + caffeine-treated (D), and caffeine-treated (E) rats are shown; scale bar = 50 μm. Representative immunohistochemical images of α-SMA in Control (F), TAA-3-Week-treated (G), TAA-6-Week-treated (H), TAA + Caffeine-treated (I), and Caffeine-treated (J) rats are shown, scale bar = 100 μm. The positive area of TGF-β is shown (K) (n = 3). The protein level of TGF-β in liver tissue samples was determined by western blot analysis (L) (n = 3), and β-actin was used as a control. The positive area of α-SMA is shown (M) (n = 3). The protein level of α-SMA in liver tissue samples was determined by western blot analysis (N) (n = 3), and β-actin was used as a control. The values are presented as fold increases in optical density values normalized to the values of the control group (control = 1). Each bar represents the mean value ± SE. (a) P < 0.05 compared with the control group; (b) P < 0.05 compared with the TAA-3-Week group; (c) P < 0.05 compared with the TAA-6-Week group.
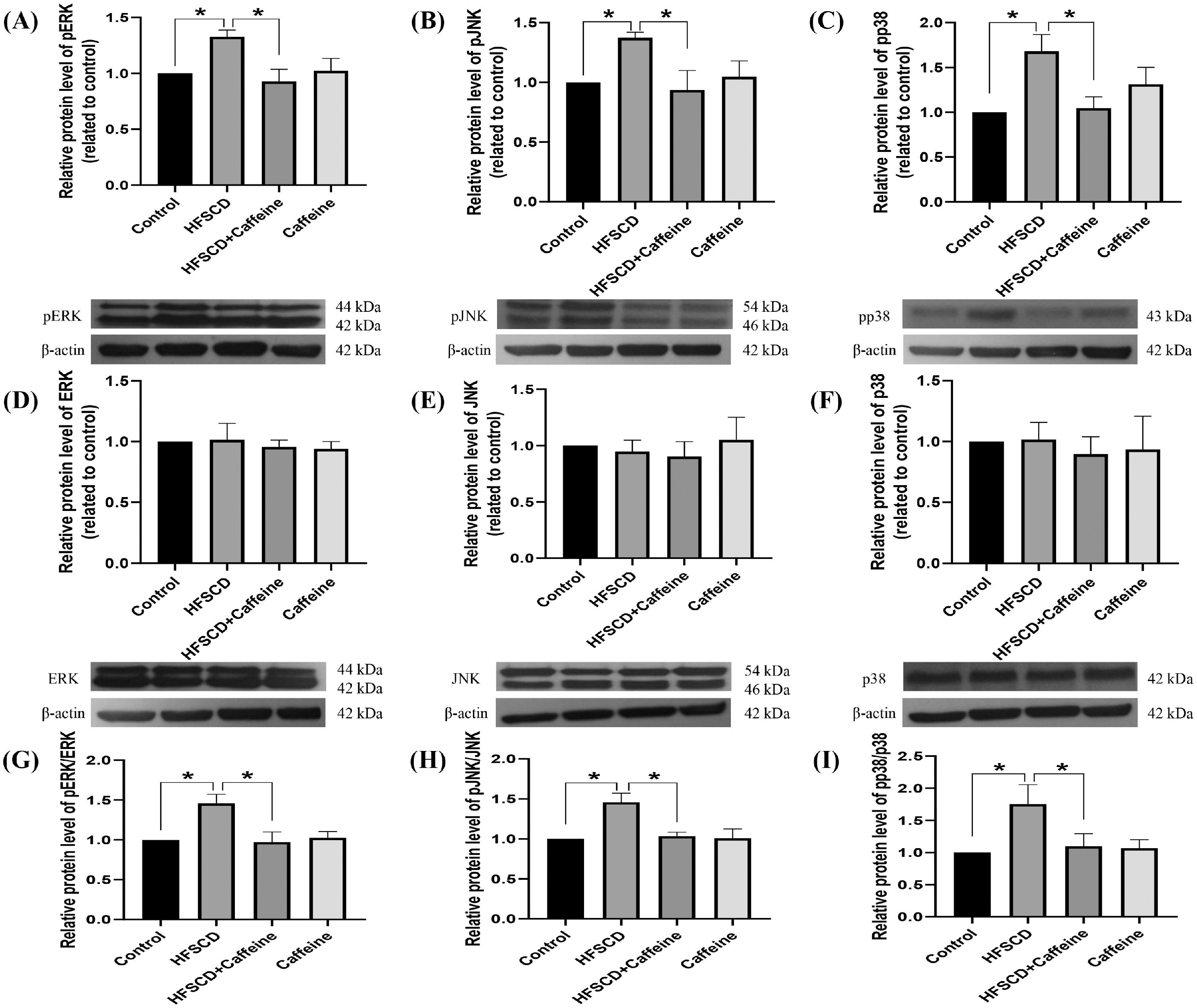
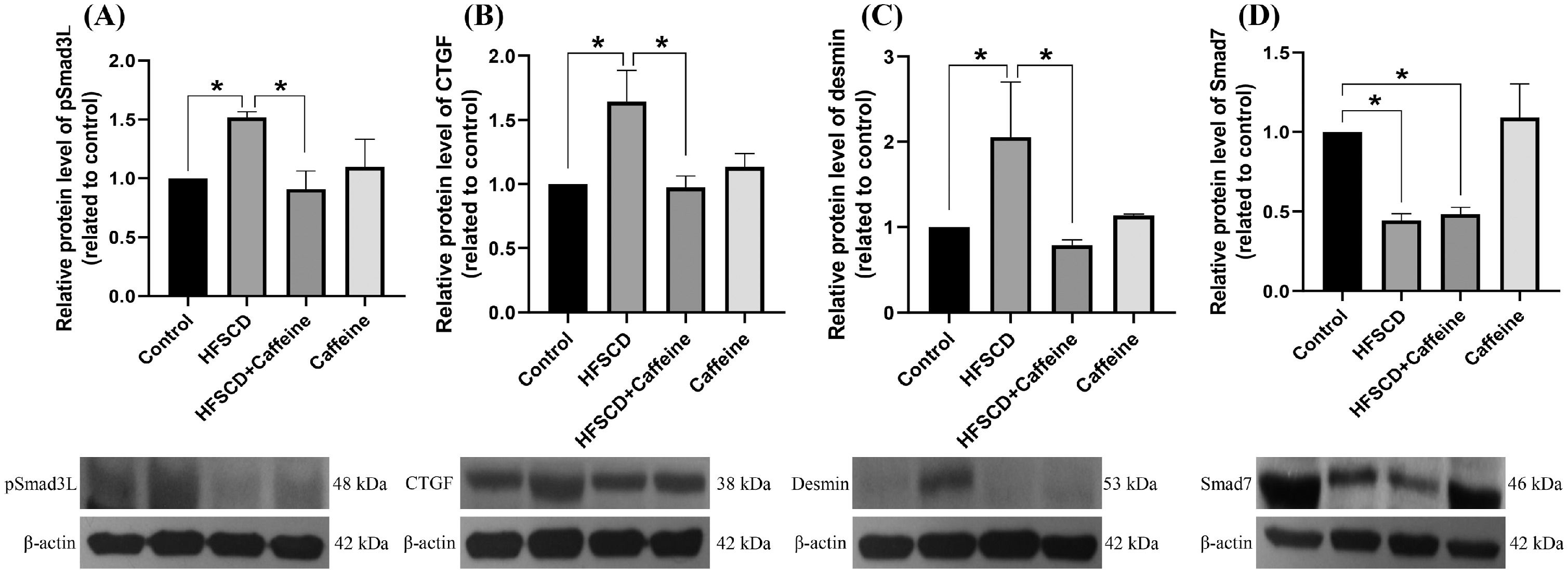
The levels of phosphorylated ERK, JNK, and p38 increased significantly with the administration of an HFSCD, while caffeine administration completely prevented these elevations (Fig. 5A-C). The total protein levels of ERK, JNK, and p38 were not affected in any of the groups (Fig. 5D-F). The pERK/ERK, pJNK/JNK, and pp38/p38 ratios increased significantly in the HFSCD group, but caffeine completely preserved these ratios to within normal levels (Fig. 5G-I). pSmad3L protein levels were increased by an HFSCD, and caffeine prevented this increase (Fig. 6A). The levels of the fibrogenesis-associated protein CTGF and desmin (an indicator of HSCs) were increased significantly in the HFSCD group; notably, caffeine preserved the normal levels of these proteins (Fig. 6B, C). The levels of Smad7, an inhibitory Smad, were significantly decreased in the HFSCD group; unfortunately, caffeine failed to prevent this decrease (Fig. 6D).
. The protein levels of pERK (A), pJNK (B), pp38 (C), ERK (D), JNK (E), and p38 (F) in liver tissue samples from the control, HFSCD, HFSCD + caffeine, and caffeine groups were determined by western blot analysis (n = 3), and β-actin was used as a control. pERK/ERK (G), pJNK/JNK (H) and pp38/p38 (I) ratios. The values are presented as fold increases in optical density values normalized to the values of the control group (control = 1). Each bar represents the mean value ± SE. (*) indicates P < 0.05.")
Caffeine prevents fibrosis by blocking the MAPK pathway in rats fed a high-fat, high-sucrose, high-cholesterol diet (HFSCD). The protein levels of pERK (A), pJNK (B), pp38 (C), ERK (D), JNK (E), and p38 (F) in liver tissue samples from the control, HFSCD, HFSCD + caffeine, and caffeine groups were determined by western blot analysis (n = 3), and β-actin was used as a control. pERK/ERK (G), pJNK/JNK (H) and pp38/p38 (I) ratios. The values are presented as fold increases in optical density values normalized to the values of the control group (control = 1). Each bar represents the mean value ± SE. (*) indicates P < 0.05.
. The protein levels of pSmad3L (A), CTGF (B), desmin (C), and Smad7 (D) in liver tissue samples from the control, HFSCD, HFSCD + caffeine and caffeine groups were determined by western blot analysis (n = 3), and β-actin was used as a control. The values are presented as fold increases in optical density values normalized to the values of the control group (control = 1). Each bar represents the mean value ± SE. (*) indicates P < 0.05.")
Caffeine prevents fibrosis by blocking the pSmad3L pathway in rats fed a high-fat, high-sucrose, high-cholesterol diet (HFSCD). The protein levels of pSmad3L (A), CTGF (B), desmin (C), and Smad7 (D) in liver tissue samples from the control, HFSCD, HFSCD + caffeine and caffeine groups were determined by western blot analysis (n = 3), and β-actin was used as a control. The values are presented as fold increases in optical density values normalized to the values of the control group (control = 1). Each bar represents the mean value ± SE. (*) indicates P < 0.05.
Histologically, NASH is characterized by steatosis and inflammatory infiltration, while fibrosis may or may not be present [11,34,35]. Fibrosis has gained much attention because several studies have shown that fibrosis is the main determinant of mortality in NASH patients [11]. In this study, we found that caffeine effectively exerted antifibrotic effects on both experimental NASH and TAA-induced chronic intoxication models. Previously, we showed that the mechanism of caffeine was associated with its antioxidant properties by increasing nuclear factor-E2-related factor-2 and decreasing the inflammatory nuclear factor-κB (NF-κB) signaling pathway (unpublished observations). Here, we show that caffeine modulates MMPs and downregulates canonical and noncanonical fibrogenic Smad signaling pathways, HSC activation and ECM deposition. Taken together, our results strongly suggest that these mechanisms act in a concerted manner to reduce fibrogenesis.
Activated HSCs are the most important producers of ECM [36]. Therefore, activation of these cells is considered a crucial step in fibrogenesis, and efforts have been made to prevent or reverse this activation to interfere with the production of fibrotic tissue as an antifibrotic therapy. There are several mechanisms by which HSCs can become activated [37,38]. Alcoholic liver disease, NASH, chronic hepatic viral infection, cholestasis, and other repetitive insults to the liver lead to the release of several cytokines that may induce HSC activation [39]. Notably, our results show that both models of liver damage induce HSC activation and that caffeine attenuates this effect. Most likely, the ability of caffeine to block HSC activation is one of the fundamental mechanisms of action of this alkaloid.
MMP activation is considered beneficial in the process of liver fibrosis because MMPs are responsible for cleaving ECM [40], eliminating the excessive accumulation of fibrotic tissue; however, there is also information that MMP activation can favor the fibrogenic process because these enzymes cleave TGF-β from its deposits in the ECM, and once free, activated TGF-β induces HSC activation, migration, proliferation and ECM deposition [41,42]. It seems reasonable that the beneficial/deleterious effects of MMP activity depend on the fibrosis stage and the type of MMP. Our results show that livers damaged by either TAA or HFSCD treatment have increased MMP activities, while caffeine attenuates this effect, leading to decreased levels of free active TGF-β, fewer activated HSCs, and reduced ECM production, suggesting that the antifibrotic activity of caffeine may be associated, at least partially, with MMP activity attenuation. Additionally, the antioxidant and immunomodulatory properties of caffeine in these models of liver damage (unpublished observations) may account for the antifibrotic effects observed in this study because free radicals may lead to inflammation, which in turn triggers fibrosis [43].
Previously, the ability of caffeine to prevent TAA-induced experimental fibrosis has been reported [17,39,40]. In those studies, caffeine was administered in combination with TAA; in contrast, in the present study, caffeine was administered to TAA-induced rats at 3 weeks to assess the ability of this alkaloid to slow the progression of fibrosis. Notably, caffeine was effective when administered to TAA-treated rats and attenuated fibrogenesis by decreasing TGF-β and α-SMA protein levels and modulating MMP activity.
Different kinases can phosphorylate Smad3 in the carboxyl-terminal to form pSmad3C (this is the case for the TGF-β receptor) or in the linker region forming pSmad3L (this phosphorylation is achieved by MAPK) [44–46]. pSmad3C is fibrogenic and is part of the canonical Smad signaling pathway; pSmad3L is mitogenic [47] and triggers the proliferation of activated HSCs to exacerbate scar tissue deposition and is considered the noncanonical pathway. In particular, activated (phosphorylated) JNK plays a pivotal role in the formation of pSmad3L and the activation and proliferation of HSCs that drive fibrogenesis, leading to advanced cirrhosis [48,49].
Novel antifibrotic mechanisms of caffeine were discovered in this study. The effect of caffeine on the inflammatory and fibrogenic MAPK and Smad3 pathways was evaluated in rats fed an HFSCD. Indeed, HFSCD feeding increased pJNK, pERK, and pp38 levels, and these factors are involved in the Smad3 and NF-κB signaling pathways [12,50]. We found for the first time that the administration of caffeine prevented the increase in these proteins in HFSCD-fed rats. The protein levels of desmin are indicative of HSCs, while CTGF and pSmad3L are important fibrogenic mediators and were increased significantly by HFSCD feeding. Notably, caffeine prevented the increases in these proteins. Smad7 inhibits the TGF-β signaling pathway; therefore, we evaluated whether caffeine acted by increasing this negative regulator. However, we found no effect on this protein. Therefore, it seems that the antifibrotic effect of caffeine is associated with the downregulation of the fibrogenic signaling pathways studied herein.
5ConclusionOur results demonstrate that caffeine attenuates experimental NASH- and TAA-induced liver damage and fibrosis progression by modulating the MAPK and TGF-β signaling pathways and decreasing HSC activation. Therefore, considering the safety profile of caffeine, it could be a potential treatment for liver diseases related to fibrosis, although more studies are needed.
AcknowledgementThe authors thank Rosa E. Flores-Beltrán, Laura Dayana Buendia-Montaño, Rafael Leyva, Benjamín E. Chavez, and Ricardo Gaxiola for excellent technical assistance. The authors also acknowledge the Animal Lab Facility to UPEAL-Cinvestav and Dr. Jorge Fernández-Hernández.
FundingThis work was supported by the National Council of Science and Technology (Conacyt) of Mexico (grant no. CF2019-53358 to P. Muriel and A1-S-27705 to V. Tsutsumi, and fellowship no. 724340 to E.E. Vargas-Pozada). Erika Ramos-Tovar thanks the postdoctoral scholarship from Conacyt.
ARRIVE guidelines statementThe authors have read the ARRIVE guidelines, and the manuscript was prepared and revised according to the ARRIVE guidelines.



