


La neuropatía óptica hereditaria de Leber es una enfermedad genética mitocondrial que típicamente produce ceguera bilateral en adultos jóvenes varones. Además de la mutación del ADN mitocondrial son necesarios otros factores genéticos y ambientales para el desarrollo de la enfermedad. En la actualidad no existe un tratamiento eficaz para la neuropatía óptica hereditaria de Leber, pero el consejo genético en portadores asintomáticos es importante. Presentamos el caso clínico de un paciente de 23 años de edad que refiere pérdida de agudeza visual central aguda unilateral que se convierte en bilateral en semanas. La exploración del fondo de ojo (tortuosidad vascular peripapilar, telangiectasias e hiperemia papilar), la angiografía con fluoresceína (con ausencia de exudación) y el engrosamiento de la capa de fibras nerviosas nos hacen sospechar la enfermedad. El test genético molecular confirma la neuropatía óptica hereditaria de Leber al encontrar la mutación G11778A en homoplasmia. A pesar del tratamiento con idebenona y suplementos vitamínicos la enfermedad evoluciona a atrofia papilar bilateral. El futuro parece estar en la terapia génica, actualmente en investigación.
Leber's hereditary optic neuropathy is a mitochondrial genetic disease that typically causes bilateral blindness in young adult males. In addition to mitochodrial ADN mutation other genetic and environmental factors are required for disease development. There is currently no effective treatment for Leber's hereditary optic neuropathy but genetic counseling is important in asymptomatic carriers. We report the case of a 23-year-old male patient with acute unilateral central visual loss that becomes bilateral in weeks. Fundus examination (peripapillary vascular tortuosity, telangiectasia and papillary hyperemia), fluorescein angiography (with no sweating) and thickening of the nerve fiber layer make us suspect the disease. Molecular genetic test demonstrate Leber's hereditary optic neuropathy mutation 11778A in homoplasmy. Despite treatment with idebenone and vitamin supplements, the disease progresses to bilateral papillary atrophy. The future appears to be in the gene therapy currently under investigation.
La neuropatía óptica hereditaria de Leber (NOHL) es una enfermedad neurooftalmológica poco frecuente producida por mutaciones en el ADN mitocondrial, con una prevalencia media aproximada en el norte de Europa de un caso por cada 40.000 personas1. Aunque fue descrita por primera vez por Theodore Leber en 1871, fue en 1972 cuando se descubrió que la herencia era mitocondrial1. La NOHL tiene penetrancia incompleta, con lo cual la existencia de la mutación no es suficiente para el desarrollo de la enfermedad.
Las mutaciones primarias más frecuentes son la G11778A, la G3460A y la T14484C, y aproximadamente un 50% de hombres y un 10% de mujeres con una mutación primaria desarrollan la enfermedad1,2. Generalmente las mutaciones son homoplasmáticas y en estos casos solo hay ADN mutante en el individuo, pero en un 15% son heteroplasmáticas. La clínica típica y la exploración nos darán el diagnóstico de sospecha, pero será el análisis genético molecular el que confirme la NOHL e identifique la mutación existente. El pronóstico generalmente es malo y no hay tratamiento curativo para esta enfermedad1. Se recomienda asesoramiento genético en los pacientes y en los portadores asintomáticos3,4.
Presentación del caso clínicoSe trata de un paciente varón de 23 años de edad, sin antecedentes personales de interés, que acude a Urgencias de Oftalmología por pérdida aguda de la visión central del ojo izquierdo (OI) de 24h de evolución. También refiere la visión más apagada de los colores, sin otra sintomatología asociada. Como antecedentes familiares destaca una prima materna diagnosticada de neuropatía óptica unilateral idiopática en seguimiento por Neurología hace unos años.
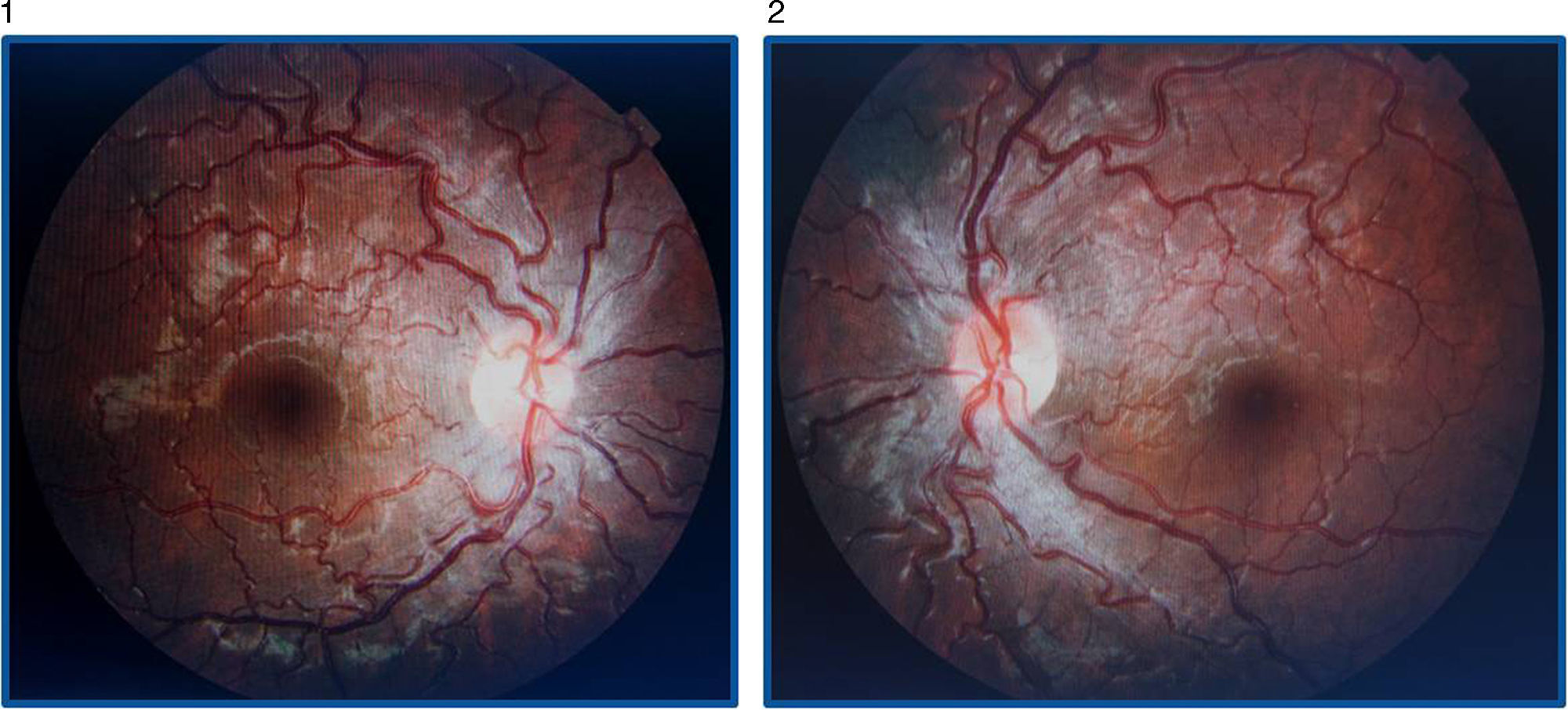
La AV (con corrección óptica de miopía y astigmatismo miópico leve) por el OI es de 0.4 y por el ojo derecho (OD) es de 1. No se observan alteraciones en el polo anterior y no hay un defecto pupilar aferente relativo. En el fondo de ojo destaca la tortuosidad vascular generalizada en ambos ojos, con hiperemia papilar bilateral y telangiectasias peripapilares, siendo el resto de la exploración normal (figs. 1 y 2).

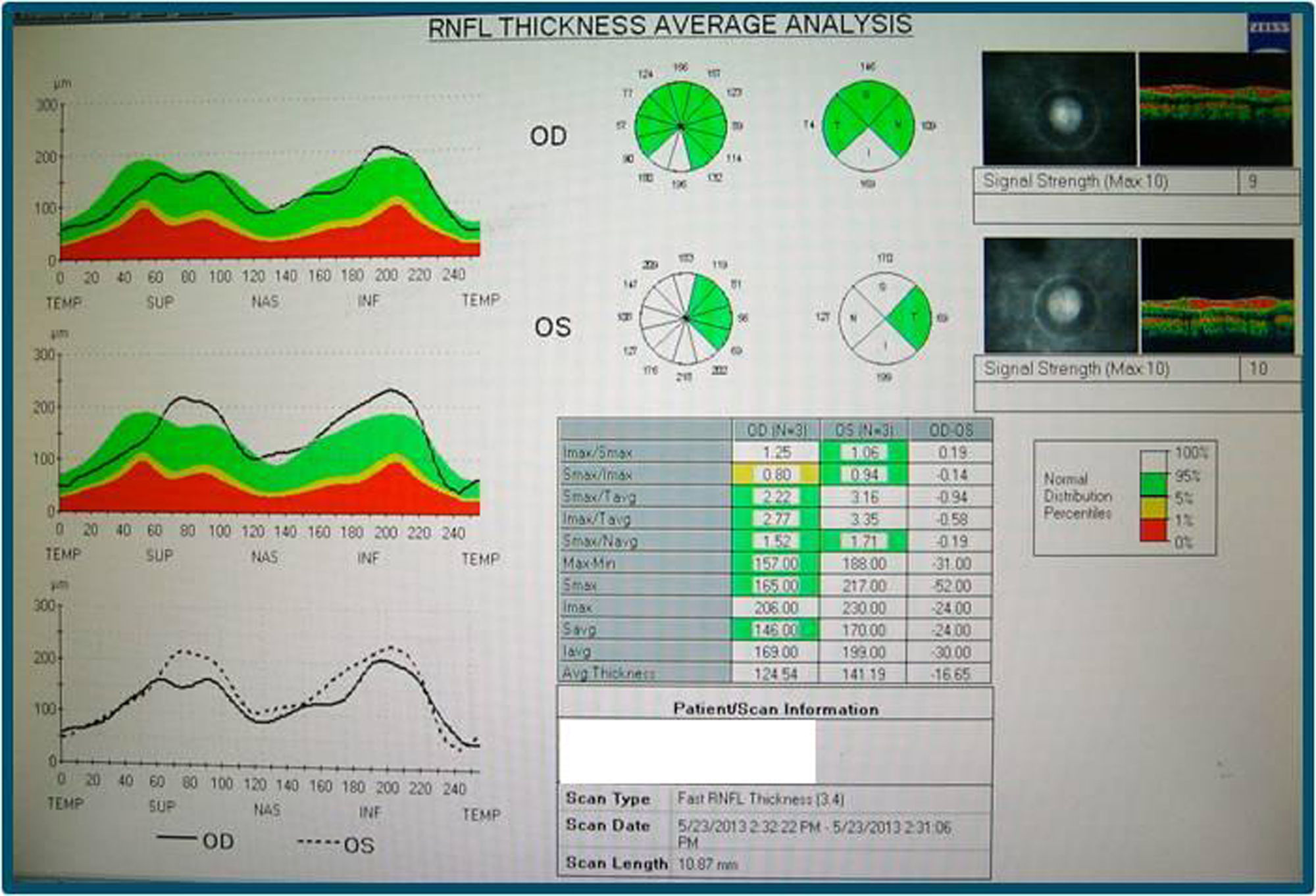
En la angiografía con fluoresceína no hay pérdida de fluoresceína (figs. 3 y 4) y la campimetría muestra un escotoma paracentral bilateral, mayor y más definido en OI. Los potenciales evocados visuales tienen aumento de la latencia y disminución de la amplitud de los potenciales en el nervio óptico izquierdo; en el derecho son normales. La tomografía de coherencia óptica (stratus time-domain) muestra un aumento del espesor de la capa de fibras nerviosas (OD: 141.29μ y OI: 124.54μ), siendo normal a nivel macular; y la tomografía axial computarizada de órbitas y cerebro fue normal.
.")
Se solicitan en conjunto con el Servicio de Neurología autoinmunidad (ANA, anticuerpos antifosfolípidos), serología (para VIH, Brucella, Borrelia y sífilis), estudio de trombofilia, RMN cerebral y orbitaria y punción lumbar, con análisis serológico y citológico del LCR, siendo todo normal.
A la semana el paciente refiere disminución de AV por el OD y la exploración a los 2 meses muestra una baja AV bilateral: en el OD de 0.2 y en el OI de cuenta dedos a medio metro.
En el fondo de ojo se observa disminución de la hiperemia papilar con palidez temporal sin telangiectasias peripapilares. La campimetría tiene un escotoma más extenso paracentral y superior bilateral y en la tomografía de coherencia óptica (stratus time-domain) persiste un grosor aumentado papilar (OD: 148.83μ, OI: 136.70μ) (fig. 3).
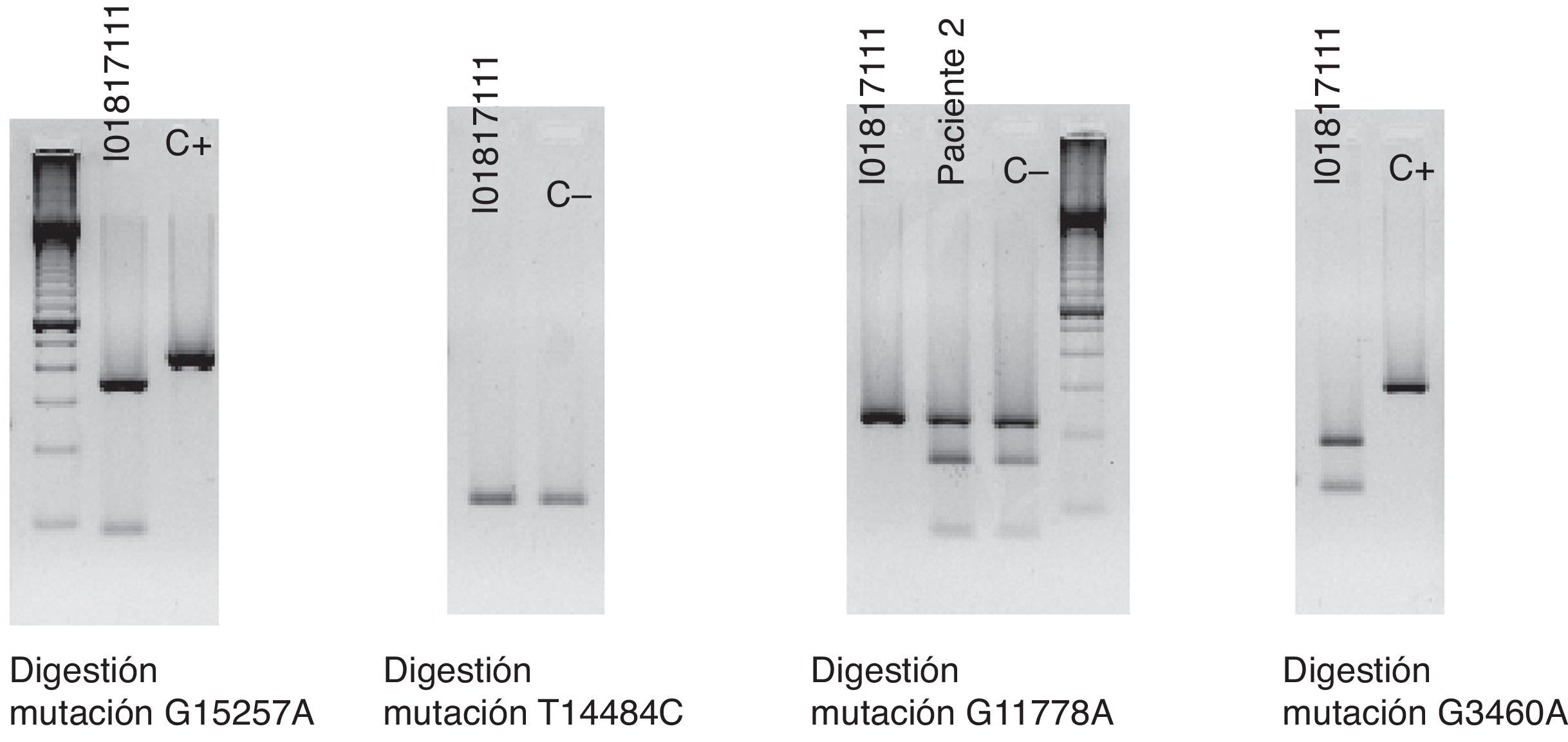
La clínica y la exploración nos hacen sospechar de una posible LHON y se solicita un test genético molecular que encuentra la mutación G11778A en homoplasmia y confirma el diagnóstico. En dicho test se descartan, a su vez, otras mutaciones involucradas en esta enfermedad, como son G15257A, T14484C y G3460A (fig. 4).

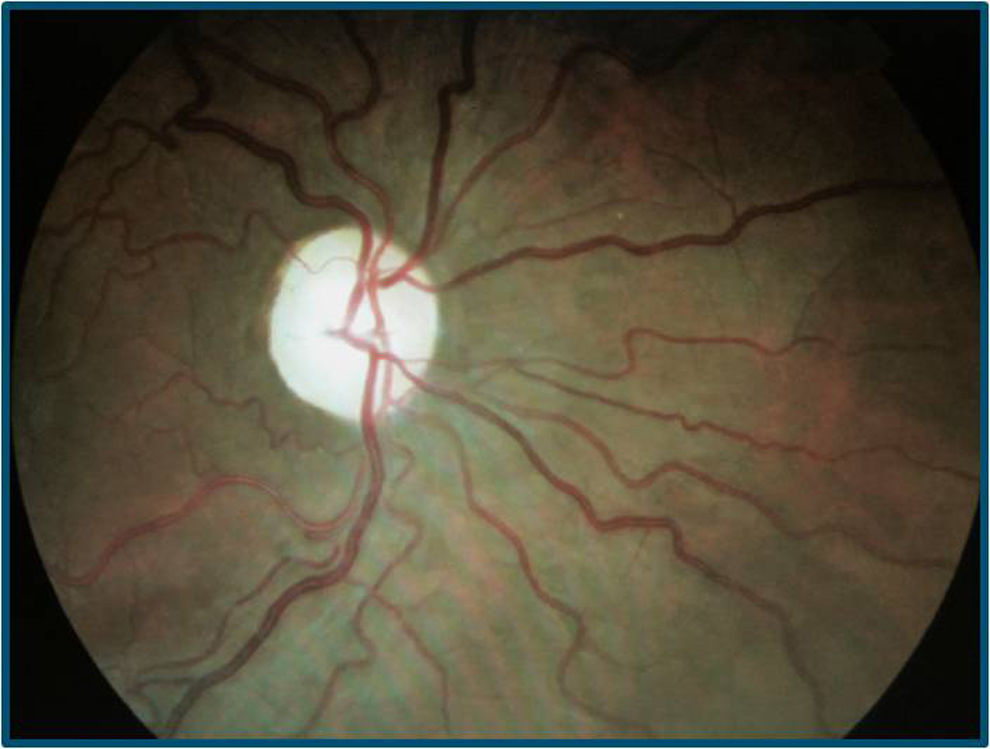
El paciente se somete a tratamiento con idebenona y suplementos vitamínicos durante un año, pero continúa la disminución de AV bilateral (en el OD de cuenta dedos a medio metro y en el OI de movimiento de manos), con evolución a atrofia papilar bilateral (figs. 5 y 6) La tomografía de coherencia óptica (cirrus spectral-domain) destaca una disminución del espesor medio de la capa de fibras nerviosas de 73 y 72μ en OD y OI, respectivamente.
Discusión

El diagnóstico de confirmación de la NOHL se realiza con el test genético molecular en sangre periférica, que tiene una fiabilidad del 100%. Como la penetrancia es incompleta, además de las mutaciones se debe tener en cuenta que otros factores genéticos y ambientales precipitan la enfermedad1.
El asesoramiento genético es difícil porque no se pueden detectar las personas en riesgo de atrofia óptica, y en caso de heteroplasmia no se puede determinar la cantidad de ADN mutante que se puede transmitir4. Lo que sí se sabe es que las mujeres transmiten la mutación a toda la descendencia y los hombres no la transmiten en ningún caso. La edad (década de 20-30 años) y el sexo (más frecuente en los hombres, 5/1) aumentan el riesgo de padecer esta enfermedad4.
En los portadores asintomáticos se ha observado en algunos casos un engrosamiento de la capa de fibras nerviosas y se cree que cuando se asocia a una disminución de la amplitud del electrorretinograma en patrón puede predecir la enfermedad2,5. El consejo genético en portadoras asintomáticas es importante para informarles de las características de esta enfermedad y de que van a transmitir la mutación a todos sus descendentes –tanto varones como hembras– y que los varones no van a transmitir la enfermedad en ningún caso. También es interesante recomendar el control de los factores externos como el tabaco o el alcohol4.
No existe ningún tratamiento eficaz para esta enfermedad. Se han usado agentes farmacológicos como la coenzima-Q (ubiquinona) o su cadena corta, idebenona, que se cree que pueden mejorar el flujo axoplásmico y prevenir el estrés oxidativo1,4,6,7. La eficacia de estos tratamientos es controvertida y se cree que pueden ser más efectivos como terapia de prevención antes de presentar la pérdida de visión.
La brimonidina1 es otro fármaco antiapoptótico agonista de los receptores alfa-2, con efecto neuroprotector de las células ganglionares de la retina, y puede ser una ayuda después de la pérdida visual para intentar conservar el otro ojo.
También se ha recomendado el uso de vitaminas como E, C, B1, B2, B12, sin evidencia demostrada de su efectividad1,4,6. La terapia génica1,5 actualmente está en investigación y parece tener un efecto prometedor en el periodo ventana con el segundo ojo sin afectación.
ConclusionesLa NOHL es una enfermedad mitocondrial sin tratamiento en el momento actual y con mal pronóstico. El asesoramiento genético no es fácil en muchos casos, pero parece razonable aconsejar a los portadores de las mutaciones primarias el control de los factores de riesgo que se creen asociados con el desarrollo de la enfermedad. El desarrollo de la investigación con la terapia génica podría ser una buena opción en el futuro.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores han obtenido el consentimiento informado de los pacientes y/o sujetos referidos en el artículo. Este documento obra en poder del autor de correspondencia.
FinanciamientoLos autores no recibieron patrocinio para llevar a cabo este artículo.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
