



Las crisis convulsivas son una complicación ampliamente descrita de la lesión cerebral aguda y de las patologías que requieren de intervención neuroquirúrgica. Los fármacos antiepilépticos se utilizan con frecuencia en la profilaxis y tratamiento anticonvulsivo en pacientes neurocríticos, siendo la primera una práctica controversial, ya que sus potenciales efectos adversos podrían afectar la evolución del paciente. La incidencia de las convulsiones varía en función de la extensión de la lesión neurológica, de su localización y de las intervenciones realizadas. Es por esto que el uso de fármacos antiepilépticos, ya sea en tratamiento o en profilaxis anticonvulsiva, debe también considerar a los nuevos fármacos incorporados al mercado, que en función de sus características farmacocinéticas, pueden representar una alternativa de similar eficacia y mayor seguridad.
El propósito de este artículo es revisar la literatura y directrices actuales, con el fin de delinear el uso adecuado de los fármacos antiepilépticos en el ámbito del cuidado neurocrítico.
Seizures are widely described as a complication of acute brain injury and in diseases that require neurosurgical intervention. Antiepileptic drug are frequently used in the prophylaxis and in the anticonvulsant therapy in neurocritical patients, the first being a controversial practice because their potential adverse effects could affect patient outcomes. The incidence of seizures varies depending on the extent of neurological damage, its location, and interventions, which is why the use of antiepileptic drug, in either treatment or anticonvulsant prophylaxis, should also consider new drugs that entered the market, because according to their pharmacokinetic characteristics, may represent an alternative of similar efficacy and greater safety.
The aim of this article is to review the literature and current guidelines, in order to delineate the proper use of antiepileptic drugs in the field of neurocritical care.
Las crisis convulsivas (CC) son una complicación frecuente luego de una lesión cerebral aguda o de una patología que requiera de una intervención neuroquirúrgica, siendo un ejemplo de estos los tumores, la hemorragia subaracnoidea y el traumatismo encefalocraneano. Estas CC, pueden llevar a un aumento de la presión intracraneana, hipoxia, e incluso a resangrado posterior a una hemorragia subaracnoidea, pudiendo verse afectado el estado neurológico del paciente y en última instancia, aumentando la morbi-mortalidad1. El tratamiento tradicional y estandarizado de las CC se basa fundamentalmente en la administración de fármacos antiepilépticos (FAEs) que incrementen el umbral crítico, tanto en situaciones agudas como a largo plazo y que de esta forma inhiban la aparición o impidan la propagación de la CC, sin modificar el sustrato patológico que genera la susceptibilidad de base. El tratamiento farmacológico de las CC y de la epilepsia presenta tres escenarios distintos: prevención primaria de las crisis convulsivas, tratamiento de las crisis convulsivas en situaciones agudas y prevención secundaria a largo plazo de las crisis epilépticas. A pesar de que la incidencia de las CC varía en gran medida según el grado de lesión neurológica, localización de la lesión, y las intervenciones realizadas, el uso de FAEs en la profilaxis anticonvulsiva es una práctica común, pero controversial. Esto, debido a que los FAEs más utilizados corresponden a los FAEs antiguos (fenitoína, ácido valproico, fenobarbital, etc.), que a pesar de representar una alternativa efectiva en el manejo y prevención de las CC, se han asociado a peores resultados debido a sus características farmacocinéticas, que determinan sus interacciones farmacológicas y hace necesaria la medición de niveles plasmáticos, a su estrecho margen terapéutico y a sus reacciones adversas2,3.
El propósito de este artículo es hacer una revisión de la literatura y de las directrices actuales, en la profilaxis y el tratamiento de las crisis convulsivas en el paciente neurocrítico y de los fármacos antiepilépticos más utilizados en este ámbito, con el fin de optimizar su uso.
PROFILAXIS ANTICONVULSIVAEn TumoresLa incidencia de crisis convulsivas asociadas a tumores supratentoriales, varía entre el 20 al 80% según el tipo de tumor (Tu Glioneuronales, incidencia del 70-80%, en especial, en pacientes con lesiones frontotemporales e insulares; gliomas de bajo grado, 60-75% en lesión frontotemporal, insular y superficial; gliomas de alto grado, 25-60% en lesión del lóbulo temporal, superficial y grado III de la OMS; meningiomas, 20-50%, principalmente por edema peritumoral; metástasis, 20-35%, asociada a melanoma y cáncer de pulmón)4,5. La literatura y las guías actuales de tratamiento no recomiendan el uso habitual de FAEs como profilaxis anticonvulsiva, por la posible incompatibilidad con el tratamiento citostático6 (ciclofosfamida, irinotecan, paclitaxel, etc)5. En caso que fuese necesaria la terapia anticonvulsiva, se deben considerar los factores individuales de cada paciente (edad, sexo, disfunción de órganos, comorbilidad y terapia concomitante) en la elección del FAE a utilizar. Como regla general, se debería evitar el uso de inductores fuertes de la CYP3A4 como la carbamazepina, oxcarbazepina, fenitoína y fenobarbital, debido a que la interacción podría comprometer la quimioterapia concurrente5. Por esta razón, la evidencia y las guías recomiendan levetiracetam y ácido valproico como FAEs de elección para las crisis focales, características en este tipo de paciente. Si la monoterapia llegara a ser ineficiente, se recomienda la terapia combinada con ambos fármacos, pudiendo ser sinérgica. Otros FAEs recomendados, pero con menor evidencia reportada, serían la lacosamida y lamotrigina4,5,7,8.
En Hemorragia Subaracnoidea por AneurismaDe las hemorragias subaracnoideas (HSA) espontáneas, el 75-80% son secundarias a la ruptura de un aneurisma cerebral, pudiendo ser único en la mayoría de los casos y múltiple en aproximadamente el 15% de ellos9,10. Actualmente, se ha establecido que la incidencia de las CC después de la HSA se encuentra entre el 6 al 18%, caracterizándose las de aparición temprana que pueden ocurrir incluso previo a la evaluación médica. Estudios retrospectivos han identificado varios factores de riesgo asociados a la aparición de CC en HSA, incluyendo aneurismas ubicados en la arteria cerebral media, el diámetro del aneurisma, presencia de hematoma intracerebral, resangrado, infarto, bajo grado neurológico e historia de hipertensión9,11. Por otra parte, la presencia de CC post HSA, se ha asociado a mayor probabilidad de resangrado, que es una de las complicaciones tempranas más importantes de esta patología, relacionándose a una morbimortalidad aproximada del 80%12,13. El uso profilactico de FAEs en HSA, es controversial, siendo el principal factor de controversia la seguridad, debido a que está relacionado a peores resultados neurológicos y a prolongación de la hospitalización. Las guías de la “American Heart Association/American Stroke Association” (AHA/ASA) y de Messé et al en el 2009, establecen que el uso de FAEs profilácticos puede ser considerado en el período post-hemorrágico inmediato o en los primeros siete días posteriores a la ruptura del aneurisma. Sin embargo, no hacen sugerencias respecto a los antiepilépticos específicos que se pueden utilizar para la profilaxis9,14. Estas mismas guías, adicionalmente recomiendan el uso de nimodipino oral en todos los pacientes con HSA, debido a la amplia evidencia en la mejora de los resultados neurológicos (evidencia clase I, nivel A)9. Este vasodilatador cerebral, al ser metabolizado vía CYP P450 3A4, se ve afectado en gran medida por inductores fuertes de estas isoenzimas como la fenitoína, que puede llegar a disminuir el AUC plasmática de nimodipino hasta en 86%15,16. Esta interacción es de alta relevancia clínica en el tratamiento de la HSA, debido al extenso data uso de nimodipino con el fin antes mencionado y de fenitoína, que históricamente ha sido el FAE más utilizado, como profilaxis anticonvulsiva en esta patología10,13,17. No obstante, la evidencia destaca los efectos adversos, la metabolización, las interacciones y los factores de seguridad en el uso de fenitoína, en especial, su asociación a deterioro funcional y cognitivo, dosis-dependiente17. Es probable que por esta razón actualmente se ha observado un aumento en el uso de levetiracetam, incluso por sobre la fenitoína18,19, pero sin la evidencia suficiente, que permitiera avalar a uno por sobre el otro13,14,17–19.
En el traumatismo encefalocraneanoUno de los riesgos inminentes del traumatismo encefalocraneano (TEC) son las crisis convulsivas post traumáticas (CCPT). Existen las de aparición temprana, que se presentan los primeros siete días de ocurrido el trauma, teniendo una incidencia del 2.1 al 16.9%20. Alrededor del 25% de los pacientes que presentan estas crisis, podrían volver a tener en los meses o años siguientes21,22. Por otro lado, las CCPT de aparición tardía son las que se presentan luego de los siete días de ocurrido el TEC, con una incidencia que va del 1.9 a más del 30%20. Aproximadamente, el 80% de los pacientes que cursa con estas crisis, podría presentar más de una crisis a lo largo de su vida. Las heridas penetrantes tienen la mayor incidencia de generar CCPT, pudiéndose presentar en aproximadamente el 50% de los pacientes23, mientras que lesiones como la fractura de cráneo, hematomas subdurales (HSD), hematomas intracerebrales (HIC), representan un incidencia de CCPT del 20 al 25%24. Es necesario evitar las crisis convulsivas post traumáticas tempranas y tardías. Sin embargo, es también importante evitar los efectos adversos y neuroconductuales de los FAEs, en especial, por su potencial toxicidad e inefectividad previniendo crisis25. Las guías actuales del manejo de TEC mencionan que usar FAEs en epilepsia o crisis convulsivas post traumáticas es necesario, sin embargo, no recomiendan el uso de estos fármacos para prevenir las CCPT tardías. Es por esta razón, que iniciar la profilaxis anticonvulsiva luego de la primera semana de ocurrido el TEC, no sería conveniente23,25.
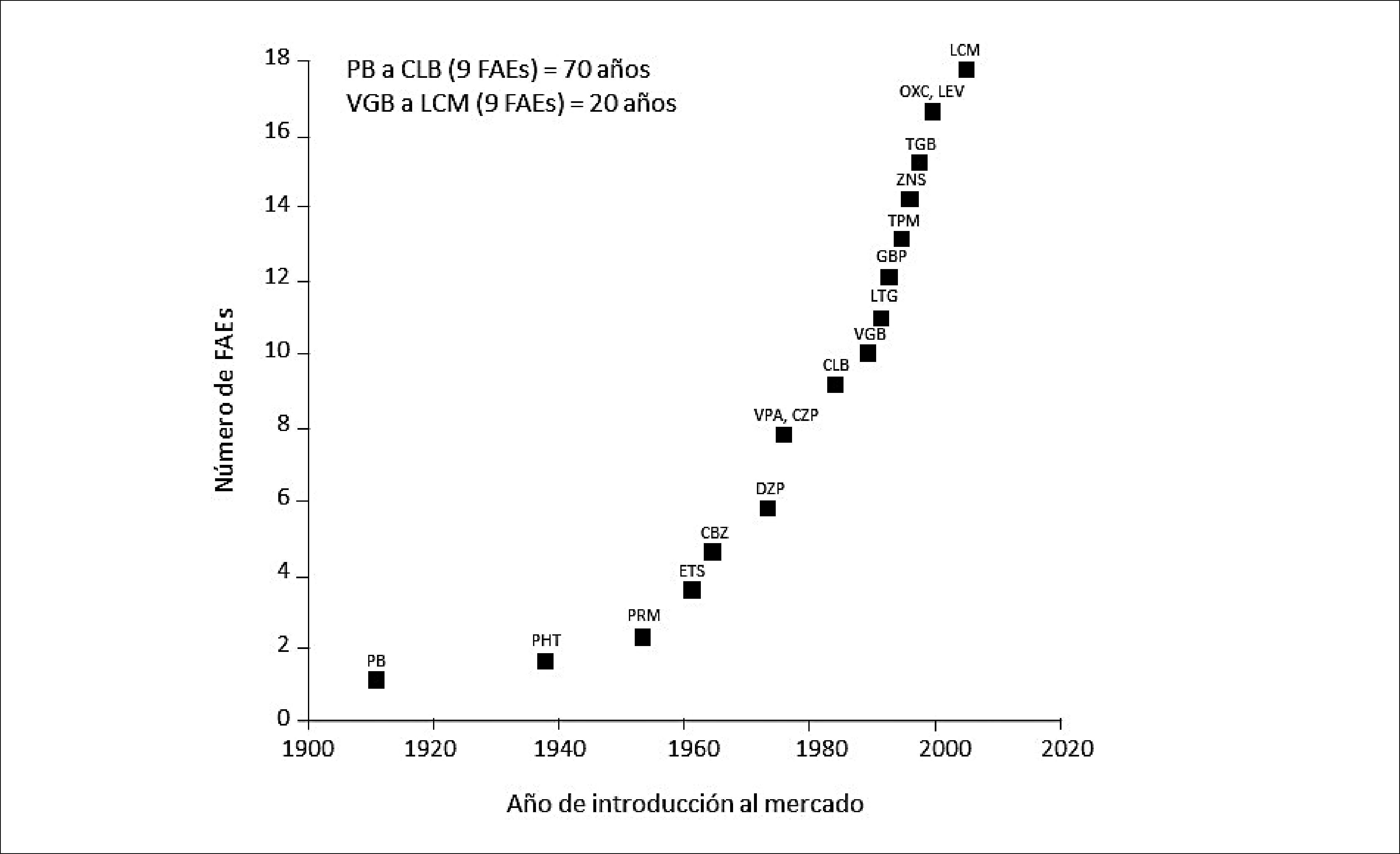
FÁRMACOS ANTIEPILÉPTICOSDebido a los múltiples efectos adversos asociados a la farmacocinética de los antiguos FAEs (reacciones idiosincráticas, reacciones adversas agudas o crónicas a nivel del SNC, interacciones farmacocinéticas/farmacodinámicas y su potencial teratogenicidad), durante las últimas décadas, ha habido un impulso sustancial en la introducción de nuevos FAEs para el tratamiento farmacológico de la epilepsia y de las CC. Este incremento se evidencia en la Figura 1, que representa la evolución cronológica de la incorporación de los FAEs a la práctica clínica, y que muestra que los primeros nueve FAEs tardaron alrededor de 70 años en introducirse al mercado, mientras que la incorporación de los nueve nuevos FAEs tardó sólo 20 años, por la necesidad de brindar mayor seguridad a la terapia anticonvulsivante26,27.

INTRODUCCIÓN DE LOS FAES A LA PRÁCTICA CLÍNICA
La introducción de los primeros nueve FAEs tardó aproximadamente 70 años, pero sólo 20 años para la introducción de los nueve FAEs siguientes. CBZ= carbamazepina; CLB= clobazam; CZP= clonazepam; DZP= diazepam; ETS= etosuximida; GBP= gabapentina; LCV= lacosamida; LEV= levetiracetam; LTG= lamotrigina; OXC= oxcarbazepina; PB= fenobarbital; PHT= fenitoína; PRM= primidona; TGB= tiagabine; TPM= topiramato; VGB= vigabatrina; VPA= ácido valproico; ZNS= zonisamida,
Adaptado de Patsalos 2000.
Dentro de los FAEs antiguos de mayor relevancia clínica, por su gran eficacia y extenso uso en el tratamiento de epilepsia, de estatus epiléptico y de profilaxis anticonvulsiva, se encuentra la fenitoína, antecedente que se respalda por la reciente declaración de la ILAE (International League Against Epilepsy) en el 2013, que ha establecido a la fenitoína dentro de los cuatro FAEs con mayor eficacia clínica, nivel de evidencia A, en el tratamiento de crisis focales28. Otros FAEs antiguos, como el ácido valproico (VPA) y el fenobarbital (PB), no cuentan con la evidencia y la efectividad necesaria para su uso en la profilaxis anticonvulsivante11,18,29,30, además de asociarse a reacciones adversas características, tales como hepatotoxicidad, trombocitopenia y neutropenia en el caso de VPA, y hepatotoxicidad, deterioro cognitivo y ataxia, asociado al uso de PB31. Por lo tanto, teniendo en cuenta estas consideraciones, es que el uso de los antiguos FAEs como la fenitoína, es controversial, debido a su cinética característica, que le confiere baja seguridad. Adicionalmente, se ha evidenciado que en unidades de paciente neurocrítico, alrededor del 43% de los pacientes podría desarrollar CC refractarias a los FAEs de primera línea (antiguos)32,33. Por esta razón, los nuevos FAEs representan alternativas de mayor seguridad, presentando cinéticas lineales, escasas interacciones y efectos adversos. Este es el caso de levetiracetam, que a pesar de contar con un par de décadas de uso clínico, también ha sido considerado por la ILAE dentro de los cuatro FAEs con mayor eficacia clínica, en el tratamiento de crisis focales28. De la misma forma, la lacosamida también representa una nueva alternativa, pero que aún requiere de experiencia y evidencia clínica para posicionarse en cuanto a su efectividad34–36.
FenitoínaDebido a la eficacia de Fenitoína (FNT) como FAE y anticonvulsivante, es que ha sido utilizada ampliamente en la práctica clínica y en unidades de paciente neurocrítico en la profilaxis y tratamiento de CC37. A pesar de esto, su seguridad es controversial, debido a sus características farmacocinéticas que le confieren una absorción variable, estrecho margen terapéutico, alta unión a proteínas plasmáticas, metabolismo saturable y numerosas interacciones farmacológicas. Luego de la administración de una dosis oral, la Cmáx se alcanza entre las 1-6h en comprimidos de liberación inmediata y a las 4-12h en las presentaciones de liberación prolongada34. La absorción se ve influenciada significativamente por la eliminación, es por esto que luego de la administración de altas dosis, su absorción se podría extender hasta las 60h38. Adicionalmente, la biodisponibilidad se ve afectada en pacientes con alimentación enteral por sonda nasogástrica, atribuyéndose una reducción de la absorción gastrointestinal de hasta el 70-80% de FNT. Aunque el mecanismo exacto de esta interacción se desconoce, se estima que existe una incompatibilidad física entre la FNT y determinados componentes en las fórmulas de alimentación enteral, que resulta en la formación de complejos (FNT-calcio, FNT-complejos proteicos)34,39. Luego de una dosis de carga, se sugiere medir niveles plasmáticos (NP) a las 2h si se realizó carga endovenosa o entre las 6-8h si se realizó carga oral para evaluar la obtención de concentraciones terapéuticas (10-20μg/mL). Una vez logrado el estado estacionario, se recomienda repetir NP si se observan cambios en el estado de enfermedad del paciente, en su tratamiento farmacológico, en la respuesta terapéutica o si se presentan síntomas/signos de toxicidad37,40. Se ha descrito que el estado estacionario, que corresponde al estado de equilibrio entre la absorción y eliminación, se logra entre las 3 a 5 vidas medias (T½) de un medicamento. A pesar de esto, y tomando en cuenta los t1/2 de FNT que varían entre las 22h para la oral y 10-15h para FNT endovenosa, se ha observado variabilidad en el tiempo para alcanzar este estado (entre 3 a 50 días) por su cinética de saturación40,41. Debido a la baja solubilidad en agua de FNT endovenosa, la formulación contiene 40% de propilenglicol, 10% de alcohol, e hidróxido de sodio para ajuste de pH. Esta solución es altamente cáustica y podría causar necrosis en los tejidos por extravasación38, por esta razón, la velocidad de administración recomendada es de 10-25mg/min, mientras que la máxima recomendada se ha establecido en 50mg/min, en especial, para minimizar los efectos cardiovasculares de la FNT (antiarrítmico clase IB, usado en la intoxicación por digoxina y en fibrilación auricular, con acción vasodilatadora periférica e inotropismo negativo) y del propilenglicol, el que podría causar bradicardia y asistolia en dosis tóxicas38,41. A pesar de que el suero fisiológico es su disolvente descrito, se recomienda no diluir, menos aún en suero glucosado, debido a la formación de cristales que generan un precipitado incoloro42. Al ser una formulación liposoluble, la FNT atraviesa rápidamente la Barrera Hemato Encefálica (BHE), alcanzando una concentración máxima (Cmáx) a los 15min38. Esto ocurre sólo con la fracción activa (10%), ya que el 90% restante, unido a albúmina, no puede atravesar la BHE41. Por su alta afinidad a proteínas plasmáticas, en pacientes en estado de hipoalbuminemia, insuficiencia renal y hepática, se ha asociado con la acumulación de compuestos endógenos que desplazan a la FNT de los sitios de unión de proteínas plasmáticas34. Por esta razón, en pacientes en estos estados, se recomienda la medición de niveles plasmáticos de FNT libre (1-2μg/mL) por sobre la medición de FNT total (10-20μg/mL), y a pesar de que este rango de FNT libre corresponde al ampliamente utilizado, se recomienda considerar el factor temperatura en esta medición, ya que la unión de FNT a proteínas disminuye significativamente al aumentar la temperatura, por ello, se han sugerido rangos de FNT libre de 0.8-1.6μg/mL para análisis realizados a temperatura ambiente, manteniéndose el rango habitual para mediciones realizadas a 37°C (temperatura fisiológica)37. En Chile, la técnica de medición de FNT libre es escasa y aunque existen fórmulas, ecuaciones (ecuación de Winter-Tozer: FNT libre estimada=((NP FNT total)/(0,2*Albúmina+0.1))*0.1)) y software de simulación farmacocinética que estiman la cantidad de FNT libre, se recomienda la medición plasmática, debido a que estas fórmulas sólo contemplan la albúmina plasmática del paciente, el NP de FNT total y el ClCr, sin considerar la competencia por la unión a proteínas plasmáticas de compuestos endógenos (ácidos grasos libres, bilirrubina y compuestos urémicos37,40 y las interacciones farmacológicas34,37. En este sentido, el ácido valproico, la tolbutamida, el ácido acetilsalicílico y algunos AINEs desplazan a la FNT de la unión de proteínas plasmáticas, pudiendo verse aumentado el porcentaje de FNT libre, si el desplazamiento además inhibe su metabolismo. FAEs inductores enzimáticos, como la carbamazepina y fenobarbital, producen efectos variables e impredecibles sobre la concentración de FNT. Por otro lado, la oxcarbazepina (>1200mg/día), topiramato y ácido valproico, pueden incrementar las concentraciones séricas de FNT34. El volumen de distribución (Vd) de FNT es de 0,7±0,1Lkg. Menos del 5% de una dosis se excreta sin cambios, y posee una alta variabilidad interindividual, debido a ser ampliamente hidroxilada, principalmente por CYP2C9 y CYP2C19, a un metabolito inactivo. La eliminación sigue una farmacocinética no lineal de Michaelis-Menten, es decir, la tasa de metabolismo disminuye al aumentar la dosis, pudiendo saturarse incluso en rangos de niveles terapéuticos40. Por lo tanto, pequeños incrementos en la dosis, pueden traducirse en aumentos desproporcionados de la concentración sérica34,35. El T½ de FNT depende de la concentración plasmática. En adultos y pacientes geriátricos con NP>10mg/L, el T½ puede variar entre 30-100h, mientras que en niños y en pacientes neurocríticos, puede ser menor a 10h34. En este contexto de la cinética errática de este FAE, se pueden observar importantes efectos adversos mayoritariamente concentración dependiente, que van desde reacciones dermatológicas (rash, síndrome Stevens-Johnson, necrólisis epidérmica tóxica, etc), hepáticas (hepatotoxicidad, Reacción a Drogas con Eosinofilia y Síntomas Sistémicos o DRESS) y neurológicas (ataxia, diplopia, nistagmus, incluso se han descrito efectos neuroconductuales con deterioros cognitivos)34,35,41.
LevetiracetamLevetiracetam (LEV) se absorbe rápidamente luego de la administración oral, obteniéndose Cmáx plasmática a las 1.3h, con una biodisponibilidad (BD) mayor al 95%. Debido a la buena BD de LEV oral, se han realizado estudios en individuos sanos, que avalan la intercambialidad de la administración oral versus la endovenosa43, lo que posibilitaría el uso de LEV oral para dosis de carga, en caso de no contar con la presentación endovenosa. La fracción de absorción no se altera en presencia de alimentos, sólo se enlentece27,34. LEV no se une a proteínas plasmáticas, y aunque los datos de su distribución en tejidos humanos no se han objetivado, se ha descrito que penetra rápidamente a líquido cefalorraquídeo (LCR) alcanzando la Cmáx a las 3-5 horas, con un Vd aparente de 0.5-0.7L/kg, el que se ve disminuido en pacientes críticos, alcanzando los 0.21L/kg (1.28). Su cinética es lineal, siendo proporcional a los rangos de dosificación establecidos (500-4000mg), sin evidencia de acumulación tras múltiples dosis. El estado estacionario se alcanza entre las 24 y 48h. A pesar de que el T½ varía entre 6-8h (5.2h en pacientes críticos1 y 10-11h en pacientes geriátricos), el intervalo de administración se ha establecido en cada 12h, debido a la afinidad por el sitio de acción, que el flujo de salida del LEV del LCR y del extracelular, observándose una prolongación de entre el 50-100% del T½. Aproximadamente el 34% del LEV es metabolizado, mientras que el 66% de la dosis se elimina inalterada en orina. Esto, ocurre principalmente por hidrólisis de la acetamida en sangre y al no ser un fármaco autoinducible, no se le atribuyen las clásicas interacciones de relevancia clínica de los FAEs (digoxina, warfarina, ACO). Sin embargo, se ha asociado a interacciones con carbamazepina y topiramato, observándose un aumento de los efectos adversos de estos, sin aumento de sus niveles plasmáticos27. El clearance renal indica una excreción por filtración glomerular y una posterior reabsorción tubular parcial, en un rango de 0.6mL/min/kg, valor que se ve aumentado en casi un 40% en pacientes críticos1,27. En pacientes con insuficiencia renal moderada a severa, se alcanza una reducción del clearance del 35-60%, y un aumento de la Cmáx en estado estacionario, en comparación con individuos sanos43. En pacientes sometidos a hemodiálisis, se ha observado una reducción del 50% de la dosis en sesiones de 4h, por lo tanto, en estos casos se recomienda la suplementación post-dialisis del 30-50% de la dosis diaria27,44. Aunque los niveles plasmáticos terapéuticos de LEV se han establecido entre los 12-46μg/mL, no se ha determinado la utilidad y relevancia clínica de su monitorización, debido a la estabilidad de su farmacocinética34. Sin embargo, se recomienda monitorizar si se observan efectos adversos, que van desde la somnolencia, alteraciones conductuales y en menor frecuencia, trombocitopenia reversible1 y nefritis intersticial27,45,46.
LacosamidaLacosamida (LCM) es un antiepiléptico sintético aprobado en el año 2008 por la FDA, que está indicado como terapia coadyudante en el tratamiento de las crisis de inicio parcial con o sin generación secundaria, en pacientes con epilepsia. Posee mecanismo de acción dual. Por un lado, favorece selectivamente la inactivación lenta de canales de sodio voltaje-dependientes, sin afectar la inactivación rápida (ms), que regula la fracción de canales de sodio disponibles para ser activados. Esta selectividad de acción, le permite estabilizar las neuronas con hiperexcitabilidad patológica, pero sin efectos sobre patrones normales de actividad neuronal repetitiva. Por otro lado, modula la actividad de la proteína mediadora de la respuesta a colapsina (collapsin-response mediator protein 2, CRMP-2), involucrada en la diferenciación neuronal, en la polarización y en el crecimiento axonal mediado por factores neurotróficos cerebrales47. LCM se encuentra entre los últimos FAEs en ser introducidos a la práctica clínica, por ende, su eficacia y seguridad aún se está evaluando. Se recomienda iniciar con dosis de 50mg dos veces al día, la que puede aumentar, según tolerabilidad, en 50mg dos veces al día cada semana, hasta una dosis máxima diaria recomendada de 400mg al día (200mg cada 12h). El retiro del medicamento debe mantener el esquema gradual de inicio (disminuir dosis en 50mg dos veces al día cada semana). Se absorbe rápida y completamente por vía oral, teniendo una biodisponibilidad aproximada del 100%, y la presencia de alimentos no altera la cantidad ni velocidad de absorción48. La Cmáx se alcanza entre las 0.5 y 4h tras la administración oral, y su absorción no es afectada por alimentos. Tras la administración intravenosa, la Cmáx se alcanza finalizada la perfusión. Estudios comparativos han sugerido utilizar dosis de carga endovenosa, y es debido a esta evidencia que en el año 2014 la EMA y la FDA aprobaron el uso de una dosis de carga de 200mg, seguido de 100mg cada 12h48–50. Su unión a proteínas plasmáticas es <15%, dializándose el 50% de la dosis administrada. Su Vd es de 0.6 L/kg, indicando una baja afinidad por tejido periférico48. El 95% se excreta por la orina, correspondiendo un 40% de la dosis a LCM inalterada y 30% a metabolito inactivo. La principal vía enzimática corresponde a CYP2C19 y cuenta con un t1/2 de 13h, el que se ve aumentado a 18-20h en pacientes con insuficiencia renal. Esto se traduce en la necesidad de ajustar la dosis en pacientes con insuficiencia renal severa, con una dosis máxima diaria autorizada por la EMA de 250mg y de 300mg por la FDA49. Se recomiendan hacer ajustes similares en pacientes con insuficiencia hepática moderada (Child-Pugh B), debido a que se ha observado un aumento del 50-60% en la exposición, por tanto, dosis máximas de 300mg al día son recomendadas en Child-Pugh B, mientras que no se recomienda su uso en pacientes con insuficiencia hepática severa. Su cinética es lineal, constante en el tiempo y tras la administración de LCM dos veces al día, el estado estacionario se alcanza a los 3 días45,47,49,51. A pesar que los niveles plasmáticos de LCM no están oficialmente establecidos (rango sugerido=5-10μg/mL; rango asociado a dosis -200 a 600mg al día =2.5-18μg/mL; nivel tóxico no establecido), se ha destacado que la concentración alcanzada en saliva es idéntica a la concentración plasmática, representando una alternativa para la medición de niveles terapéuticos48. Se ha observado que la coadministración con FNT y carbamazepina, disminuyen las concentraciones plasmáticas de LCM, lo que podría sugerir que su clearance aumenta en presencia de FAEs inductores enzimáticos36. En el contexto de una alternativa como profilaxis anticonvulsiva en el paciente neurocrítico, está descrito que la LCM interacciona con Nimodipino, pudiendo aumentar el riesgo de la prolongación del PR, el bloqueo aurículo-ventricular, y la bradicardia32,36,48. En cuanto a los efectos adversos de LCM, se ha observado desde somnolencia, n usea, mareos, diplopia, visión borrosa, vómitos, hasta prolongación del intervalo PR en el ECG y bloqueo aurículo ventricular32.
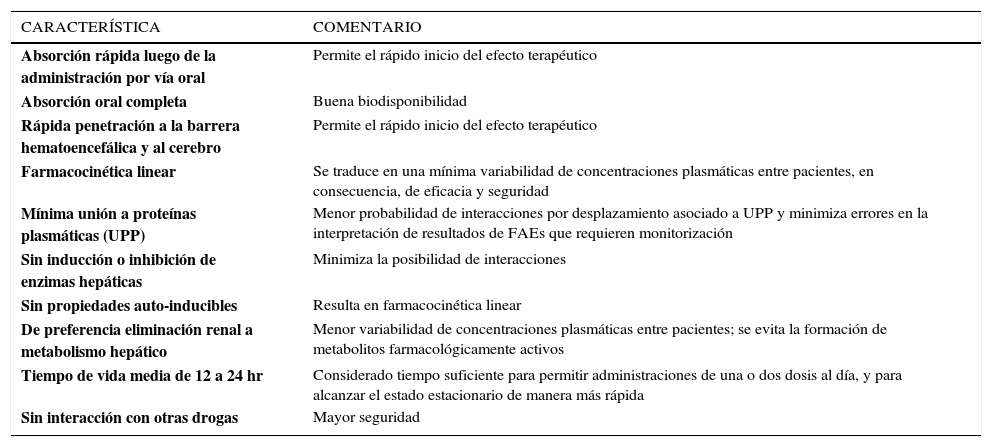
CARACTERÍSTICAS FARMACOCINÉTICAS IDEALES DE LOS FAESLas características farmacocinéticas de un medicamento determinan su uso clínico, la seguridad y su tolerabilidad. El paciente neurocrítico, presenta una mayor probabilidad de desarrollar CC por el tipo de lesión, requiriendo un tratamiento o una profilaxis eficaz con FAEs. Al mismo tiempo, estos pacientes presentan alteraciones dinámicas que determinan la farmacocinética (alteración en Vd, en ClCr, polifarmacia, aumento de PIC, etc), y por ende, la respuesta terapéutica. De esta manera, durante la última década, se han identificado numerosas características farmacocinéticas ideales en relación a los FAEs (Tabla 1).
CARACTERÍSTICAS FARMACOCINÉTICAS IDEALES DE UN FÁRMACO ANTIEPILÉPTICO
| CARACTERÍSTICA | COMENTARIO |
|---|---|
| Absorción rápida luego de la administración por vía oral | Permite el rápido inicio del efecto terapéutico |
| Absorción oral completa | Buena biodisponibilidad |
| Rápida penetración a la barrera hematoencefálica y al cerebro | Permite el rápido inicio del efecto terapéutico |
| Farmacocinética linear | Se traduce en una mínima variabilidad de concentraciones plasmáticas entre pacientes, en consecuencia, de eficacia y seguridad |
| Mínima unión a proteínas plasmáticas (UPP) | Menor probabilidad de interacciones por desplazamiento asociado a UPP y minimiza errores en la interpretación de resultados de FAEs que requieren monitorización |
| Sin inducción o inhibición de enzimas hepáticas | Minimiza la posibilidad de interacciones |
| Sin propiedades auto-inducibles | Resulta en farmacocinética linear |
| De preferencia eliminación renal a metabolismo hepático | Menor variabilidad de concentraciones plasmáticas entre pacientes; se evita la formación de metabolitos farmacológicamente activos |
| Tiempo de vida media de 12 a 24 hr | Considerado tiempo suficiente para permitir administraciones de una o dos dosis al día, y para alcanzar el estado estacionario de manera más rápida |
| Sin interacción con otras drogas | Mayor seguridad |
Adaptado de Patsalos 200427.
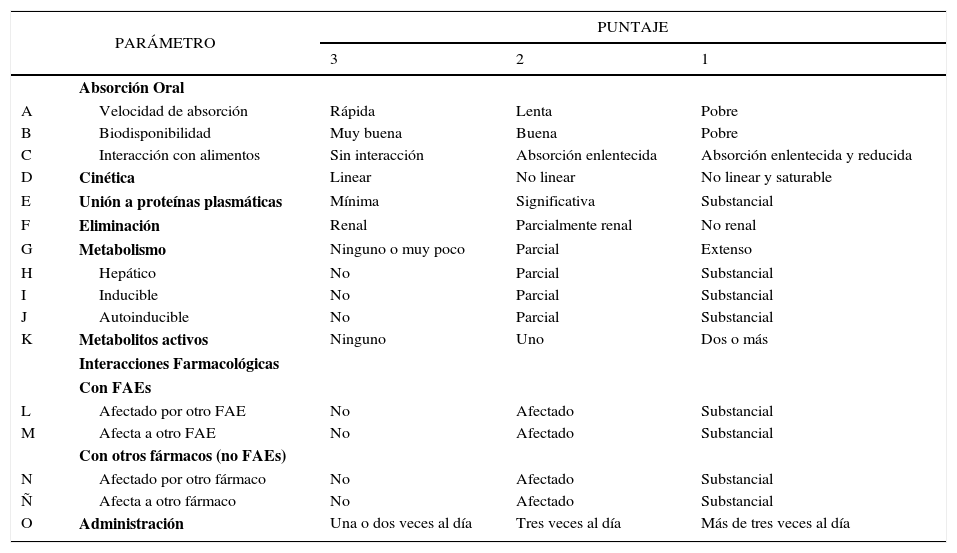
Con el fin de permitir la comparación directa entre los FAEs antiguos y nuevos, Patsalos diseñó en el año 2000 un sistema de clasificación farmacocinético semicuantitativo, el cual, permite evaluar la seguridad de los principales FAEs, considerando 16 parámetros farmacocinéticos, A-O (Tabla 2)27.
CARACTERÍSTICAS FARMACOCINÉTICAS IDEALES DE UN FÁRMACO ANTIEPILÉPTICO
| PARÁMETRO | PUNTAJE | |||
|---|---|---|---|---|
| 3 | 2 | 1 | ||
| Absorción Oral | ||||
| A | Velocidad de absorción | Rápida | Lenta | Pobre |
| B | Biodisponibilidad | Muy buena | Buena | Pobre |
| C | Interacción con alimentos | Sin interacción | Absorción enlentecida | Absorción enlentecida y reducida |
| D | Cinética | Linear | No linear | No linear y saturable |
| E | Unión a proteínas plasmáticas | Mínima | Significativa | Substancial |
| F | Eliminación | Renal | Parcialmente renal | No renal |
| G | Metabolismo | Ninguno o muy poco | Parcial | Extenso |
| H | Hepático | No | Parcial | Substancial |
| I | Inducible | No | Parcial | Substancial |
| J | Autoinducible | No | Parcial | Substancial |
| K | Metabolitos activos | Ninguno | Uno | Dos o más |
| Interacciones Farmacológicas | ||||
| Con FAEs | ||||
| L | Afectado por otro FAE | No | Afectado | Substancial |
| M | Afecta a otro FAE | No | Afectado | Substancial |
| Con otros fármacos (no FAEs) | ||||
| N | Afectado por otro fármaco | No | Afectado | Substancial |
| Ñ | Afecta a otro fármaco | No | Afectado | Substancial |
| O | Administración | Una o dos veces al día | Tres veces al día | Más de tres veces al día |
Adaptado de Patsalos 200427.
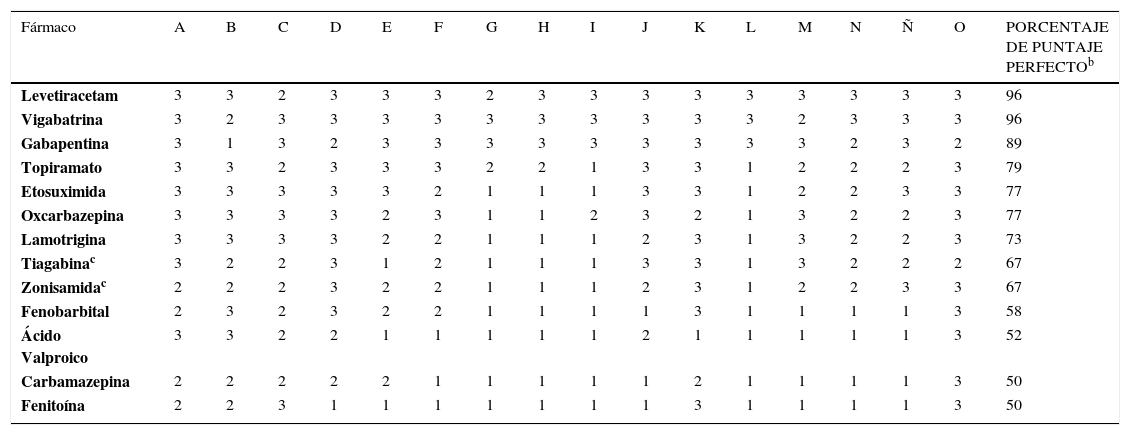
Una puntuación de 3 es óptima y corresponde a las características farmacocinéticas ideales identificadas en la última década, tal como se indica en la Tabla 3. Una puntuación de 2 es satisfactoria y de 1, insatisfactoria. El uso de este sistema, permite comparar la farmacocinética de los distintos FAEs, asignándoles un puntaje (Tabla 3). Para cada FAE, el porcentaje de la puntuación perfecta se calcula mediante la suma de los valores de cada categoría y dividiendolo por la máxima puntuación (48 puntos). Mientras mayor es el porcentaje de la puntuación perfecta, mayor seguridad representará el FAE según sus características farmacocinéticas. De esta forma y en base a esta propuesta, LEV correspondería al FAE con mayor porcentaje de puntaje perfecto en este sistema de clasificación, traduciéndose en un perfil farmacocinético ideal, mientras que carbamazepina y FNT estarían en último lugar, con 50%. Debido a que esta clasificación se realizó antes de que LCM contara con mayor evidencia clínica, Patsalos en el 2000 no la incluyó en la evaluación, sin embargo, si se extrapolara esta medición a los parámetros farmacocinéticos de LCM, podría obtener un porcentaje aproximado de 90% (puntaje de cada parámetro: 3, 3, 3, 3, 3, 3, 2, 2, 3, 3, 2, 2, 3, 2, 3, 3), debido a su amplia metabolización hepática y a su interacción con otros fármacos y FAEs.
PUNTUACIÓN DEL PERFIL FARMACOCINÉTICO DE DIVERSOS FÁRMACOS ANTIEPILÉPTICOS CON LICENCIA RECIENTE A NIVEL MUNDIALa
| Fármaco | A | B | C | D | E | F | G | H | I | J | K | L | M | N | Ñ | O | PORCENTAJE DE PUNTAJE PERFECTOb |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Levetiracetam | 3 | 3 | 2 | 3 | 3 | 3 | 2 | 3 | 3 | 3 | 3 | 3 | 3 | 3 | 3 | 3 | 96 |
| Vigabatrina | 3 | 2 | 3 | 3 | 3 | 3 | 3 | 3 | 3 | 3 | 3 | 3 | 2 | 3 | 3 | 3 | 96 |
| Gabapentina | 3 | 1 | 3 | 2 | 3 | 3 | 3 | 3 | 3 | 3 | 3 | 3 | 3 | 2 | 3 | 2 | 89 |
| Topiramato | 3 | 3 | 2 | 3 | 3 | 3 | 2 | 2 | 1 | 3 | 3 | 1 | 2 | 2 | 2 | 3 | 79 |
| Etosuximida | 3 | 3 | 3 | 3 | 3 | 2 | 1 | 1 | 1 | 3 | 3 | 1 | 2 | 2 | 3 | 3 | 77 |
| Oxcarbazepina | 3 | 3 | 3 | 3 | 2 | 3 | 1 | 1 | 2 | 3 | 2 | 1 | 3 | 2 | 2 | 3 | 77 |
| Lamotrigina | 3 | 3 | 3 | 3 | 2 | 2 | 1 | 1 | 1 | 2 | 3 | 1 | 3 | 2 | 2 | 3 | 73 |
| Tiagabinac | 3 | 2 | 2 | 3 | 1 | 2 | 1 | 1 | 1 | 3 | 3 | 1 | 3 | 2 | 2 | 2 | 67 |
| Zonisamidac | 2 | 2 | 2 | 3 | 2 | 2 | 1 | 1 | 1 | 2 | 3 | 1 | 2 | 2 | 3 | 3 | 67 |
| Fenobarbital | 2 | 3 | 2 | 3 | 2 | 2 | 1 | 1 | 1 | 1 | 3 | 1 | 1 | 1 | 1 | 3 | 58 |
| Ácido Valproico | 3 | 3 | 2 | 2 | 1 | 1 | 1 | 1 | 1 | 2 | 1 | 1 | 1 | 1 | 1 | 3 | 52 |
| Carbamazepina | 2 | 2 | 2 | 2 | 2 | 1 | 1 | 1 | 1 | 1 | 2 | 1 | 1 | 1 | 1 | 3 | 50 |
| Fenitoína | 2 | 2 | 3 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 3 | 1 | 1 | 1 | 1 | 3 | 50 |
Los parámetros farmacocinéticos abreviados en los encabezados de columna se describen en la Tabla 2
A partir de la revisión de la literatura primaria y de las directrices actuales en los principales estados patológicos observados en las unidades de pacientes neurocríticos, se evidencia que los FAEs convencionales usados en la profilaxis y tratamiento de las CC, se asocian a peores resultados debido a las interacciones medicamentosas, a la necesidad de medición de niveles plasmáticos, y las reacciones adversas. En base a las características farmacocinéticas de los nuevos FAEs, diseñados con el objetivo de brindar un mejor perfil de seguridad a la terapia anticonvulsivante, se observa que estos han representado una alternativa en pacientes neurocríticos. Sin embargo, se estima necesario contar con mayor evidencia y experiencia clínica en su uso, para poder diseñar estrategias futuras que promuevan tratamientos efectivos y seguros.
Los autores declaran no tener conflictos de interés, en relación a este artículo.