





Si bien aún la mayoría de los pacientes con hipoacusia se sigue beneficiando con los audífonos convencionales, las prótesis auditivas implantables han evolucionado vertiginosamente, existiendo en el presente una gran variedad de ellas.
Los implantes cocleares siguen siendo los más usados y en las que se tiene mayor experiencia. Las prótesis de conducción ósea implantables o semi implantables cambiaron el manejo de las atresias y malformaciones de oído externo y medio. Pese a lo prometedor que se visualiza el presente y futuro con el uso de estos dispositivos, siempre se debe tener presente que requieren de un acto quirúrgico para su implantación y que no están exentas de complicaciones, por lo cual se debe elegir juiciosamente la prótesis a usar.
While still most patients with hearing loss continue to benefit from conventional hearing aids, implantable hearing devices have rapidly evolved existing in the present a great variety of them.
Choclear implants remain the most used and in which there is greater experience. implantable or semi Implantable bone conduction prosthesis changed the management of outer and middle ear atresia and malformations. Despite how promising present and future is visualized with the use of these devices, we should always keep in mind that they require a surgical procedure for implantation and are not exent of complications, which should judiciously choose the prosthesis to be used.
Las malformaciones otológicas responden al desarrollo incompleto o a una modificación de una o más estructuras del oído durante la gestación.
Las disgenesias auditivas, también llamadas atresia aural congénita o síndrome microtia atresia, incluyen a una serie de dismorfias del pabellón auricular, del conducto auditivo externo (CAE), la membrana timpánica y el oído medio.
Epidemiología:Las disgenesias auditivas son defectos congénitos que se presentan en 1 de cada 10000 a 15000 recién nacidos vivos, excluidas las orejas en asa que constituyen una variante anatómica. Es más frecuente que ocurra en un solo oído, 80% de los casos, con cierta preponderancia en el varón y en el oído derecho.
RESEÑA EMBRIOLÓGICADesde el punto de vista embriológico, el oído externo, medio e interno derivan de las tres hojas blastoméricas.
El ectodermo somático interviene en la génesis del oído externo e interno, constituyendo los elementos epiteliales del pabellón, del CAE, la capa externa de la membrana timpánica y el laberinto membranoso del oído interno (ectodermo neural).
El mesodermo participa en la formación de los músculos y cartílagos auriculares del oído externo, los huesecillos, los músculos del martillo y del estribo, la capa media de la membrana timpánica, el laberinto perióstico y la cápsula interna del oído interno.
El endodermo contribuye en el desarrollo del oído medio, formando el sistema tubotimpánico de celdillas aéreas, desde la trompa de Eustaquio, la capa interna de la membrana timpánica, el epitelio de la caja del oído medio, y de las celdillas mastoideas.
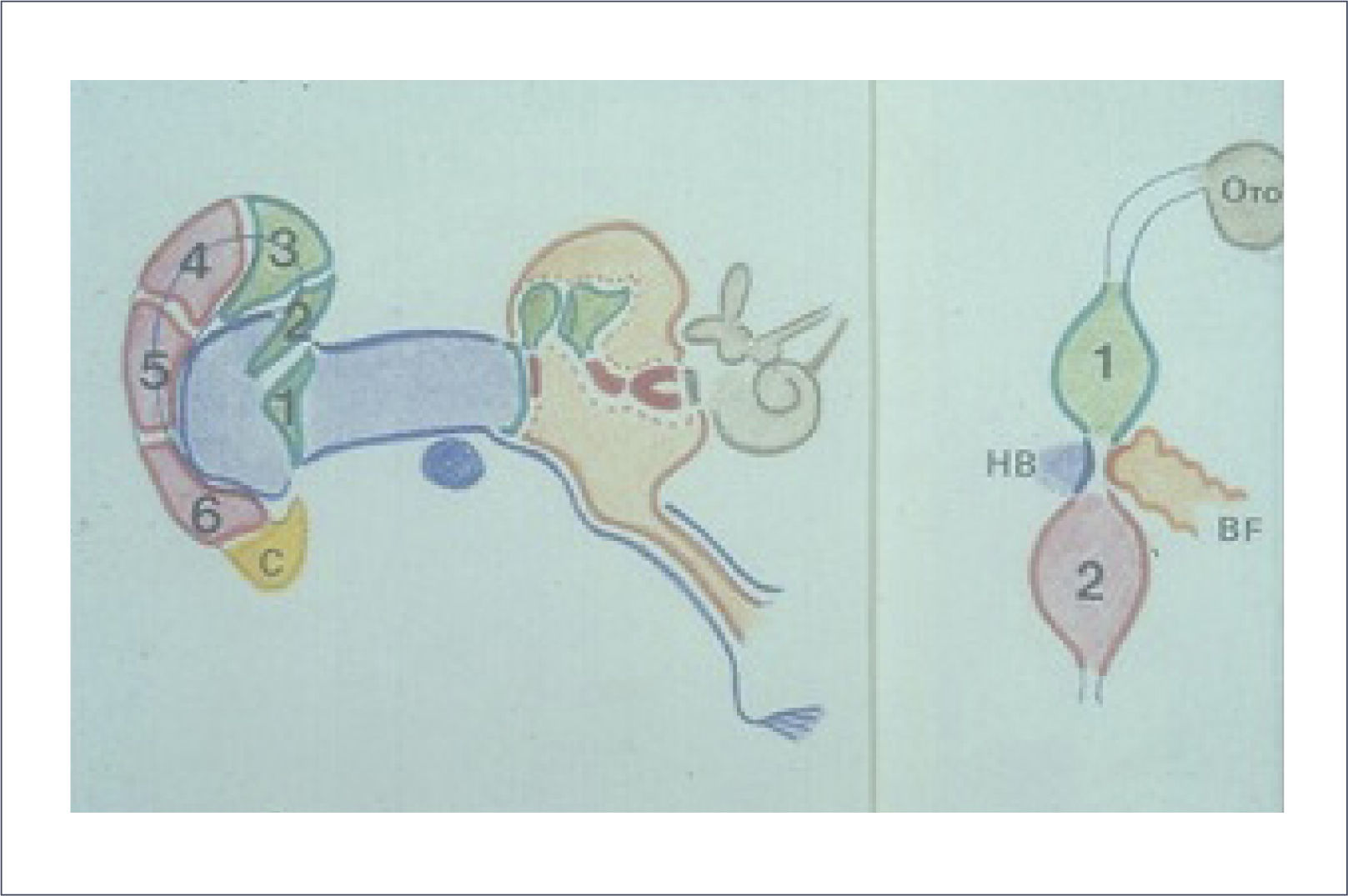
El oído externo y medio derivan de los dos primeros arcos branquiales, de la primera hendidura ectodérmica y del primer saco endodérmico según la teoría de Wood y Jones (Tabla 1); en cambio, el oído interno deriva de la vesícula ótica (Figura 1).
TEORÍA DE WOODS Y JONES
| Pabellón | Osículos | |
|---|---|---|
| 1° ARCO BRANQUIAL | Trago - Raíz del helix | Cabeza de martillo |
| Helix superior | Cuerpo del yunque | |
| 2° ARCO BRANQUIAL | Helix inferior | Apófisis larga del martillo |
| Antitrago | Apófisis descendente del yunque | |
| Concha auricular | Las cruras del estribo y la cara timpánica de la platina (la cara vestibular se origina del otocisto) | |
| 1° HENDIDURA BRANQUIAL | Conducto auditivo externo | |
| 1° BOLSA FARÍNGEA | Trompa de eustaquio-caja timpánica-ático-antro y celdas mastoideas | |
Verde: 1° Arco branquial. Rojo: 2° Arco Branquial. Azul: 1° Hendidura Branquial. Naranja: Bolsa faríngea. Marrón: Otocisto Neural

Diferentes noxas pueden generar una detención o malformación durante la embriogénesis del oído externo, medio o interno principalmente en el período comprendido entre la tercera y décima semana de gestación. Estas alteraciones pueden ocurrir debido a una embriopatía infecciosa por rubeóla, citomegalovirus o toxoplasma gondii, metabólica como el hipotiroidismo o cretinismo endémico, tóxica por la ingesta de drogas ototóxicas como la isotretinoina, talidomida, aminoglucósidos o alcohol, genética, aislada o asociada a un síndrome polimalformativo, o de causa indeterminada.
La atresia del CAE se debería a la ausencia de reabsorción del tapón meatal, o a un hiperdesarrollo del cartílago de Reichert (segundo arco branquial). Las fijaciones congénitas del estribo, aisladas o asociadas a otras malformaciones osiculares, podrían deberse a la ausencia de diferenciación del ligamento anular de la platina. Las malformaciones del martillo y del yunque pueden tener origen en una alteración de la diferenciación del cartílago de Meckel (primer arco branquial) dando lugar a una malformación de los huesecillos, o a una fijación anómala del martillo y yunque. En algunos casos, se encuentra una persistencia de la arteria estapedial.
Muchas malformaciones del oído son de origen genético, en relación con la mutación de un gen o de la transmisión de genes de expresividad variable. La transmisión sería en la mayoría de los casos de tipo autosómico dominante con penetrancia variable, aunque se han descripto casos de transmisión recesiva o ligada al cromosoma X. La mayor parte de los genes causales intervienen en la regulación de la embriogénesis craneofacial, como en el caso del síndrome de Treacher-Collins (gen TCOF1). Presentan también un origen genético y anomalías morfológicas del oído interno, el síndrome de Pendred de transmisión autosómica recesiva, el síndrome de Waardenbur y el síndrome branquio-oto-renal, estos últimos dos de herencia autosómica dominante. El síndrome branquio-oto-renal presenta también malformaciones del oído medio y externo.
En las malformaciones unilaterales, como en el síndrome de Goldenhar (displasia oculoauriculovertebral), el origen genético no está claramente demostrado. Una teoría clásica refiere que las anomalías del desarrollo en los casos unilaterales podrían ser consecuencia de una isquemia hística resultante de la obliteración de la arteria estapediana durante la embriogénesis.
CLASIFICACIÓN• Según el grado de dismorfismo del hueso timpanal (clasificación otoscópica y fonográfica).Estenosis: La reabsorción parcial del tapón meatal en la etapa gestacional, genera un CAE de estrechez variable (con riesgo de colesteatoma por descamación epidérmica). La membrana timpánica está presente, pero es hipoplásica.
Atresia: Se presenta un hueso timpanal amorfo y macizo por la falta de reabsorción central. El CAE y la membrana timpánica ausentes.
Agenesia: Sin desarrollo del hueso timpanal, del CAE y la membrana timpánica; el oído medio generalmente es hipoplásico, a expensas de un hipotímpano pequeño y con un nervio facial en su tercera porción más antepuesta y lateralizada.
• Según la forma de presentación.Puro: Compromete solo el pabellón y hueso temporal por alteración del primer y segundo arco branquial. Es la forma de presentación más frecuente.
Microsomía hemifacial: Del lado de la disgenesia la hemicara es hipoplásica.
Síndrome: La disgenesia auditiva se encuentra asociada a otras alteraciones en cráneo, paladar, columna, manos, riñón, entre otros. Algunos ejemplos son la disostosis mandibulofacial (síndrome de Treacher Collins), síndrome oculoauriculovertebral (síndrome de Goldenhard), síndrome branquio-oto-renal, disostosis craneofacial (síndrome de Crouzon), síndrome de Apert (acrocéfalo sindactilia), disostosis acrofacial (Síndrome Najer) y cromosomopatías asociadas a malformaciones del primero y segundo arco branquial como: trisomía 18 o Síndrome de Edwards y la trisomía 21 o síndrome de Down.
• Según el grado de dismorfia del pabellón auricular.Asociado a distintos grados de estenosis o atresia del CAE.
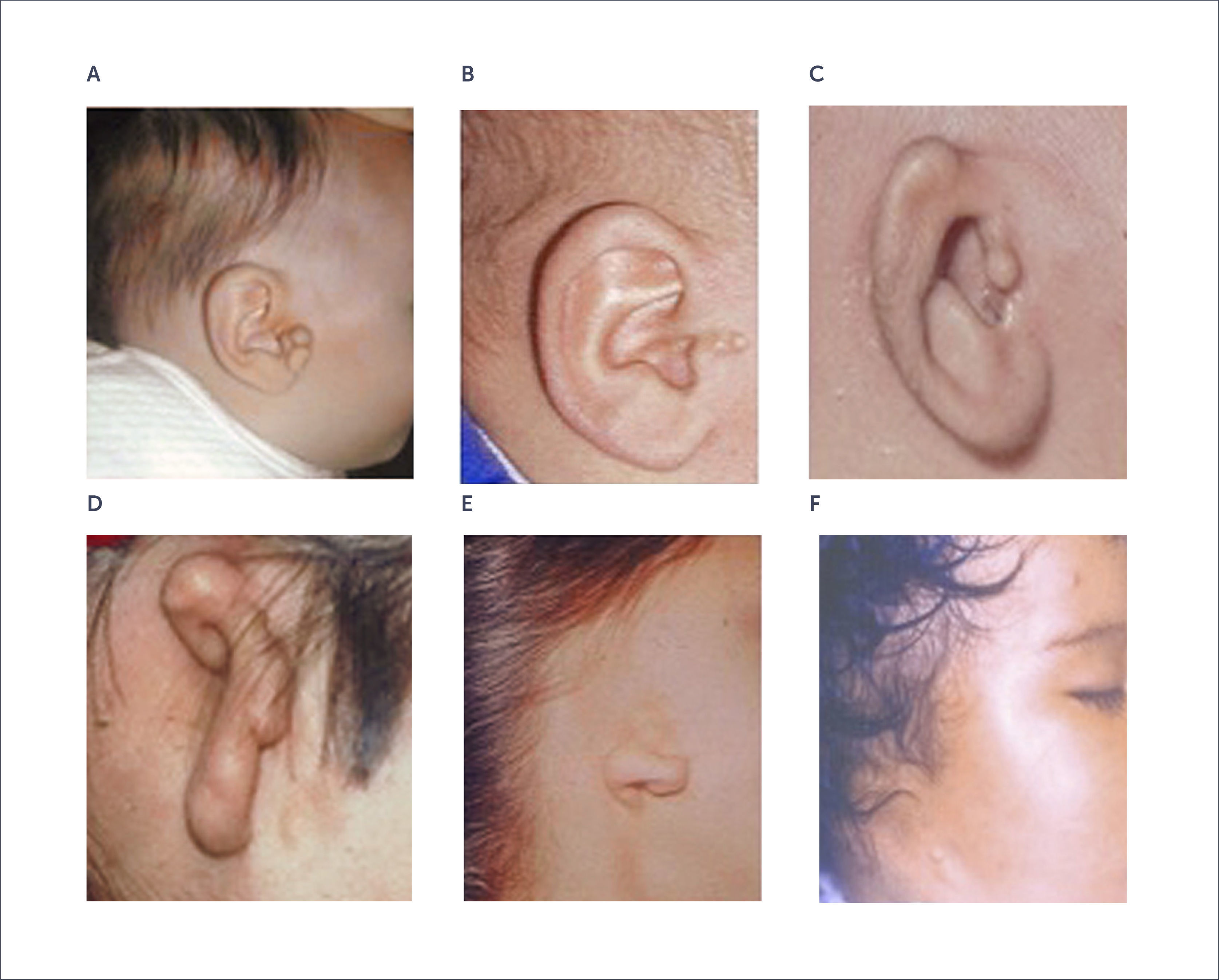
Estigmas: Pabellón auricular de características normales, la única alteración es la presencia de apéndices o mamelones cutáneo cartilaginosos, o fístulas pretragales (Figura 2 A).
 Mamelones preauriculares; B) Microtia de 1° grado; C) Microtia de 2° grado; D) Microtia de 3° grado; E) Sólo lóbulo; F) Anotia.")
Microtia de 1° grado: Pabellón ligeramente más pequeño con estructuras de forma conservada, o presenta mínimas alteraciones, como un helix superior plegado o escafa ausente (Figura 2 B).
Criptotia: Es una rara malformación que se caracteriza por la invaginación del polo superior del hélix bajo una porción de piel de la región temporal craneal.
Microtia de 2° grado: Pabellón auricular de menor tamaño y plegado sobre si mismo (oreja en canoa o caracol) (Figura 2C).
Microtia de 3° grado: Presenta un rodete cutáneo cartilaginoso en forma de maní (Figura 2 D).
Sólo lóbulo: Presencia de una formación cutánea (lóbulo) con ausencia de los seis mamelones cartilaginosos que conforman el esqueleto del pabellón (Figura 2 E).
Anotia: Ausencia completa de la oreja, se presenta en forma excepcional (1). (Figura 2 F)
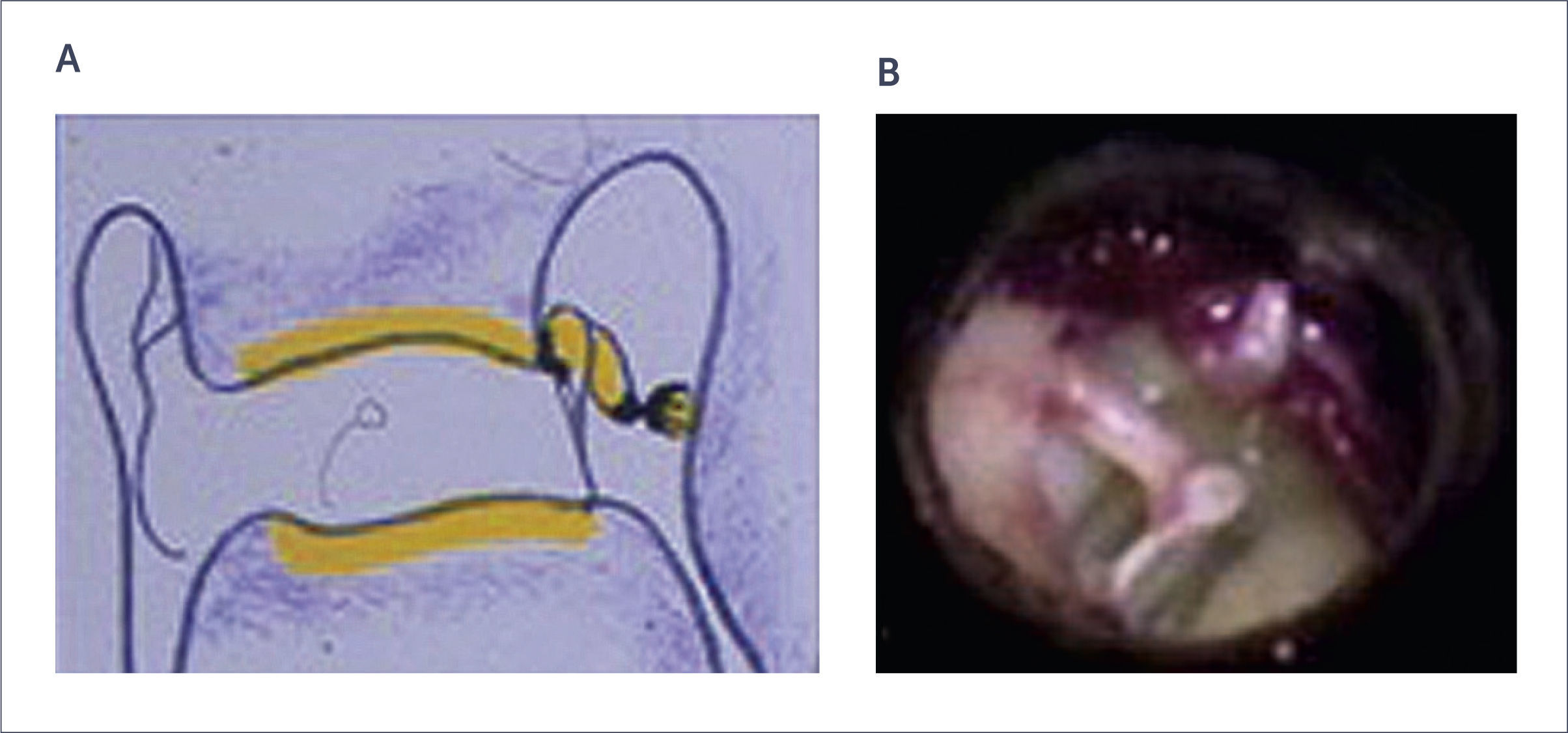
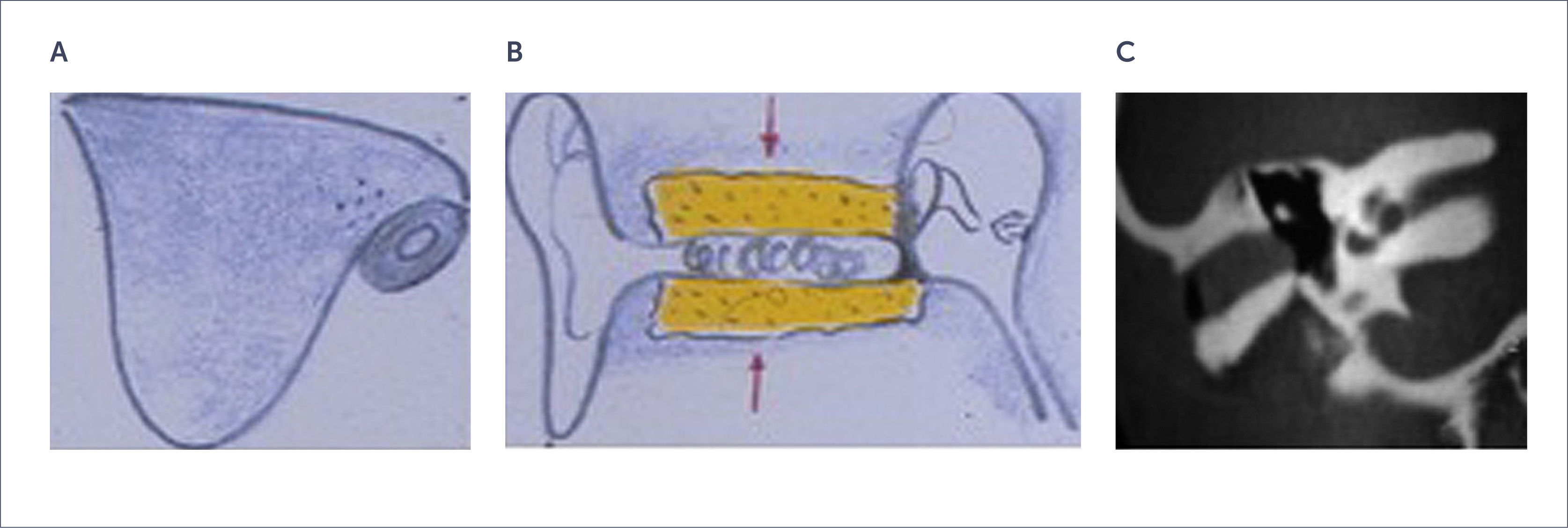
• Según el grado de malformación del hueso temporal Malformaciones menoresGrado I: Micromalformación de la cadena osicular (fijación de la cabeza del martillo, ausencia de la apófisis descendente del yunque, ausencia de la supraestructura del estribo o estribo fijo); puede asociarse a algún estigma del pabellón auricular (Figura 3).
 Esquema con CAE normal B) Ausencia de la descendente del yunque.")
Grado II: Estenosis leve del CAE, membrana timpánica hipoplásica, y micromalformacion del mango del martillo o adherencia del mismo a la pared anterior del protímpano. La cavidad del oído medio usualmente es de tamaño normal o más pequeña. (Figura 4).
Malformaciones mayores: y B) Esquema de hipertrofia del hueso timpanal. C) Imagen otoscópica con fibra óptica, muestra (1) mango de martillo fijo a la pared anterior del CAE, (2) barra del martillo. D) Malformación del martillo.")
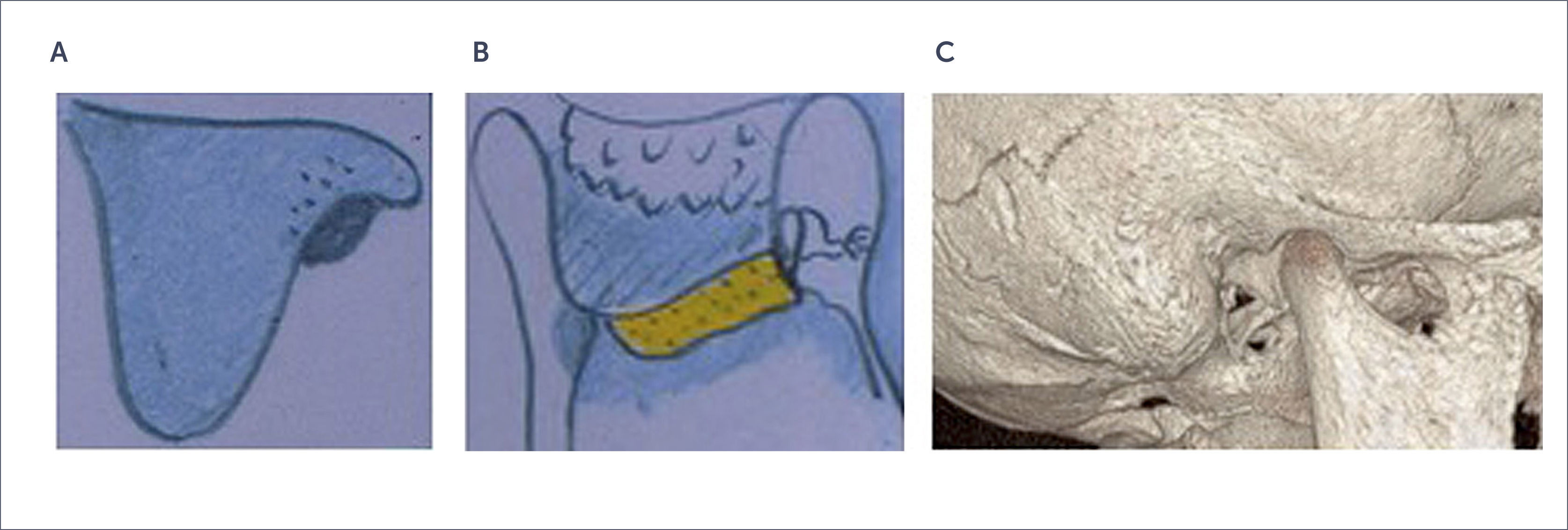
Grado III: Estenosis del CAE severa (diámetro menor de 2 o 3mm). Puede asociarse a colesteatoma del CAE por descamación y retención de queratina en la primera infancia. (Figura 5).
 y B) Hipertrofia del hueso timpanal con reabsorción central parcial; C) Estrechez del CAE. y ocupación con densidad de partes blandas (colesteatoma).")
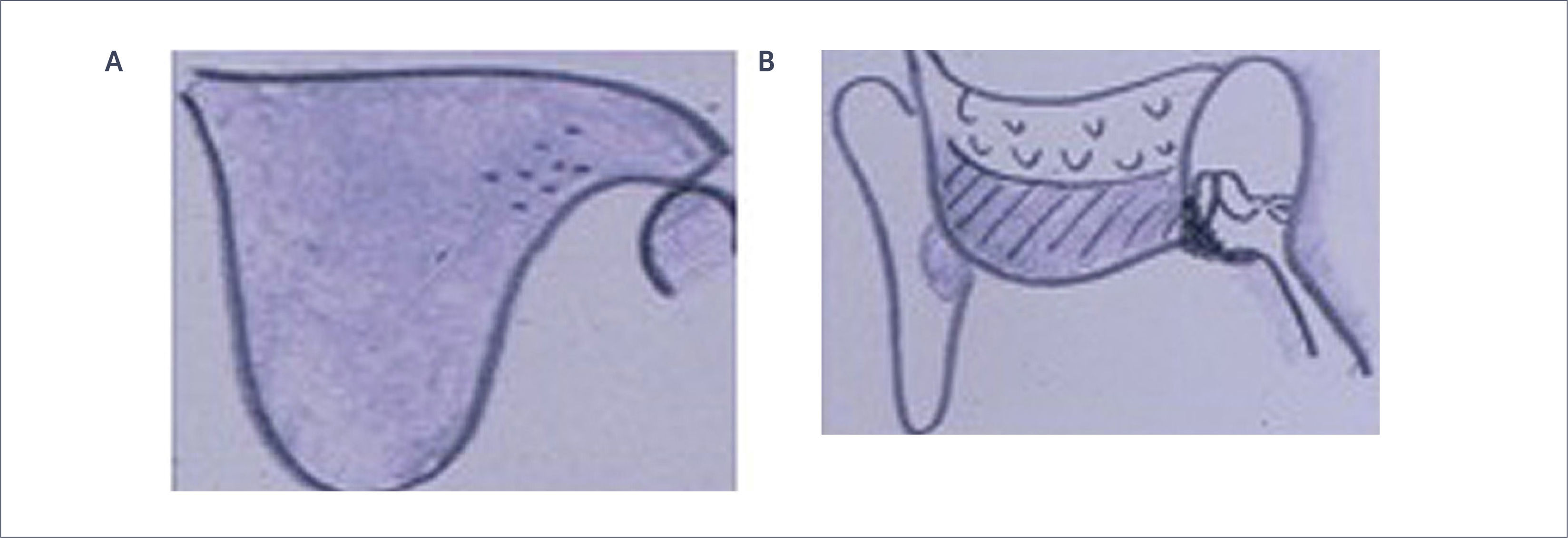
Grado IV: Hueso timpanal amorfo y compacto con cierre total del CAE óseo. (Figura 6).
 y B) Esquema con hueso timpanal amorfo y compacto; C) Corte sagital de tomografía en 3D, muestra hueso timpanal compacto, no canalizado.")
Grado V: Ausencia de hueso timpanal, agenesia del CAE óseo con distintos grados de hipoplasia del oído medio. (Figura 7).
DIAGNÓSTICO DE MALFORMACIONES DE OÍDO y B) Esquema con agenesia del hueso timpanal.")
Es prioritario tanto para el pediatra como para el médico otorrinolaringólogo realizar un examen físico integral y completo a fin de definir si se trata de un cuadro puro o sindrómico, en este último caso se debe solicitar la evaluación por genetista.
Examen Físico:- •
Pabellón auricular: observar su forma, estructuras, implantación, estigmas (fístulas, apéndices o mamelones).
- •
Meato y CAE.
- •
Tímpano: El tímpano será hipoplásico en los casos de disgenesias de 2° y 3°.
- •
Articulación temporomandibular y rama ascendente del maxilar inferior.
- •
Cráneo: evaluar aspecto y conformación de las suturas.
- •
Cara: evaluar asimetrías, hipoplasias del maxilar superior o inferior, apertura bucal, hendiduras palatinas o fisura submucosa.
- •
Cuello, tórax y extremidades: presencia de quistes branquiales, evaluar características del cuello, tórax y miembros superiores e inferiores.
Los pacientes con malformación del oído externo y/o medio presentan hipoacusia de tipo conductivo no mayor a 60db; cuando se acompañe de compromiso del oído interno la hipoacusia será mixta y sensorioneural de severa a profunda si sólo afecta al laberinto.
- 1.
Otoemisiones acústicas: posibles en caso de CAE permeable,o para evaluar el oído contralateral a la disgenesia.
- 2.
Potenciales evocados auditivos de tronco con búsqueda de umbrales por vía aérea o por vía ósea.
- 3.
Pruebas conductuales, como audiometría a campo libre o por juego según la edad del niño.
- 4.
Timpanometría e impedanciometría en conductos permeables o en el oído contralateral a la disgenesia para determinar posibles malformaciones de la cadena, en oídos aparentemente normales.
Se solicita tomografía computada de peñascos con cortes finos axiales y coronales y de alta resolución. Permite valorar el hueso temporal y el timpanal, evaluando la mastoides, la caja timpánica, su relación con el nervio facial, la cadena de huesecillos, y la conformación del laberinto óseo. La tomografía multislice permite realizar la reconstrucción en 3D de la cadena osicular, determinando la existencia de malformaciones o alteraciones en la articulación incudo-estapedial.
La tomografía se realiza alrededor de los 5 a 6 años, o antes en caso de disgenesia bilateral o en los casos de sospecha de colesteatoma por retención en conductos con un grado severo de estenosis (menor de 3mm).
GUÍA DE SEGUIMIENTO Y TRATAMIENTO EN LAS DISGENESIAS DE OÍDO SEGÚN EDADDisgenesia bilateral:Primeros meses de vida:
- •
Test audiológicos de tipo conductual.
- •
Potenciales evocados auditivos por vía aérea en caso de CAE permeable o por vía ósea.
- •
Indicar auxilio auditivo por vía ósea de uso externo (vincha ósea convencional o diadema del dispositivo osteointegrado (BAHA-SOPHONO), en caso de malformaciones mayores u otoamplífono por vía aérea en caso de malformaciones menores.
1 a 5 años:
- •
Incentivar el uso intensivo de dispositivo por vía ósea rotándolo en ambos lados, o empleando dos procesadores en caso de malformaciones mayores.
- •
Estimulación auditiva con fonoaudióloga.
- •
Controles periódicos de los umbrales auditivos.
- •
Monitoreo del desarrollo del lenguaje.
- •
Tomografía computada si se sospecha colesteatoma por retención en caso de estenosis de CAE severa.
5 a 6 años:
- •
Solicitud de tomografía computada de peñascos de alta resolución.
- •
Auriculoplastía, según requerimiento del paciente y familia, a partir de los 8 años.
- •
Atresioplastía con fines funcionales o para el uso de otoamplífonos por vía aérea.
- •
Otras alternativas para habilitación auditiva: implantes de conducción ósea.
Primeros meses de vida:
- •
Test audiológicos de tipo conductual.
- •
Potenciales evocados auditivos por vía aérea en caso de CAE permeable, o por vía ósea con umbrales.
- •
Otoemisiones acústicas, impedanciometría y reflejo acústico del supuesto oído normal a fin de descartar una micromalformación de cadena.
- •
Indicar audífono en caso de hipoacusia conductiva, mixta o neurosensorial del oído sin malformación del pabellón o conducto, sobre todo si se comprueba alteración del lenguaje.
- •
Si los estudios determinan una audición normal del oído no disgenético no requerirá dispositivo auditivo.
1 a 5 años
- •
Controles periódicos de los umbrales auditivos.
- •
Monitoreo del desarrollo del lenguaje.
- •
Tomografía computada si se sospecha colesteatoma por retención en caso de estenosis de CAE severa en el oído disgenésico.
- •
Indicar dispositivo de transmisión ósea (vincha ósea convencional o diadema del dispositivo osteointegrado) en caso de alteración del lenguaje.
5 a 6 años:
- •
Solicitud de tomografía computada de peñascos de alta resolución en las disgenesias de 3° y 4°.
- •
Auriculoplastía electiva a partir de los 8 años.
- •
Atresioplastía con fines funcionales o para el uso de otoamplífonos por vía aérea en caso de mal desarrollo del lenguaje o trastornos del aprendizaje a nivel escolar, solo en casos de muy buena puntación tomográfica.
- •
Otras alternativas para habilitación auditiva: Implantes de conducción.
En el tratamiento de las disgenesias auditivas se deben considerar dos aspectos: el estético, reparativo del pabellón auricular y el funcional auditivo, y en este último caso si la malformación es unilateral o bilateral.
Reconstrucción del pabellón auricular por auriculoplastía:A partir de los 8 años de edad. Existen distintas técnicas.
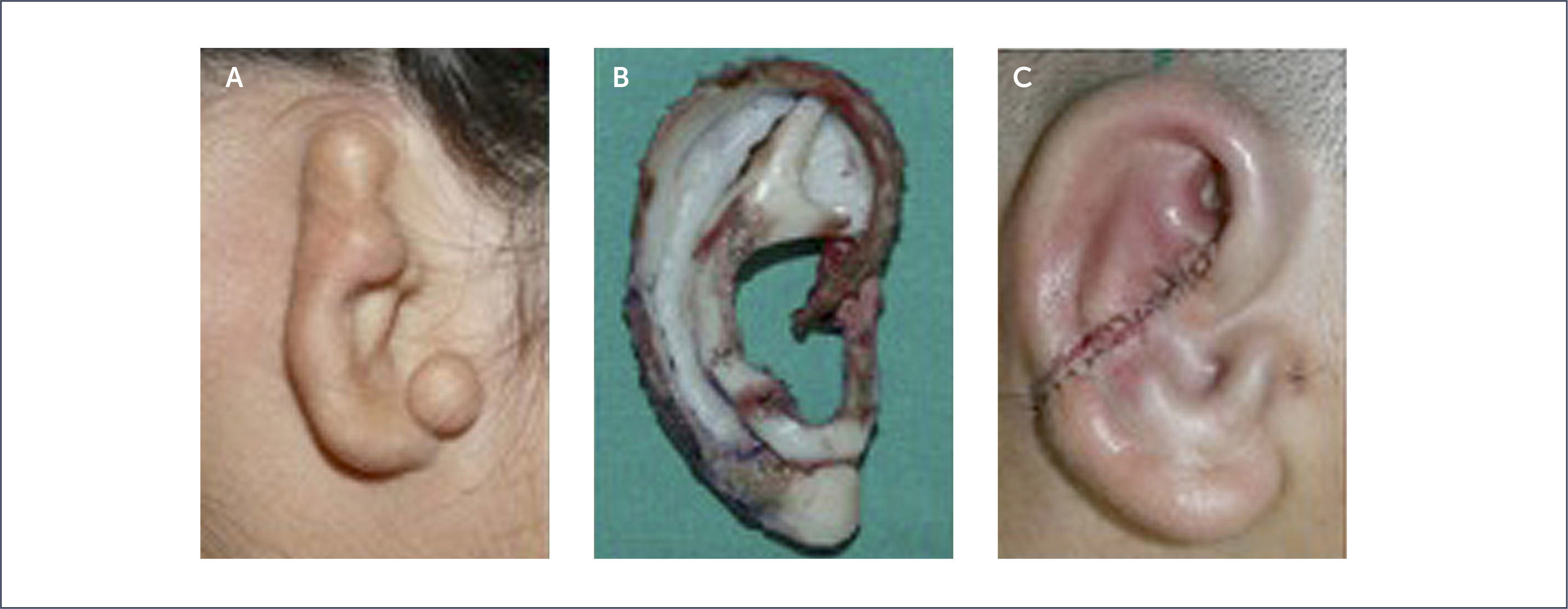
Reconstrucción con cartílago costal: es la más utilizada a nivel mundial, requiere la obtención de cartílago de la 6ª, 7ª y 8ª costillas con preservación en el tórax del pericondrio medial. Requiere tres tiempos quirúrgicos. (Figura 8).
 Microtia grado 3; B) Molde y tallado de cartílago. C) Reconstrucción con cartílago costa, foto cortesía Dra. Fernanda Valotta.")
Reconstrucción con cartílago de concha de oreja contralateral: se utiliza la técnica de Davis – Cianflone, solo para casos unilaterales. Se realiza en tres tiempos, zona dadora es el cartílago de la concha auricular contralateral. Primer tiempo, inclusión del marco óseo tallado con cartílago costal. Segundo tiempo es el despegue del pabellón con injerto retroauricular de piel total obtenido generalmente del pliegue inguinal. Tercer tiempo, reposición del lóbulo. Este tiempo puede incluirse en el segundo tiempo.
Reconstrucción con material protésico aloplástico: se han empleado distintos tipos de materiales, actualmente se utiliza silicona porosa (Medpore®) que se incluye en un bolsillo subdérmico. Permite buena resolución estética, pero es dura al tacto y puede producir isquemia del colgajo, infecciones y/o rechazo por reacción de cuerpo extraño con la consiguiente extrusión.
Orejas de siliconas externas (epítesis de pabellón): se emplea una prótesis de silicona que es anclada al hueso temporal con tornillos de titanio osteointegrados en uno o dos tiempos quirúrgicos.
CIRUGÍA CORRECTIVA DE LA ATRESIOPLASTÍASe basa en la creación de un neoconducto, se indica con el fin de otorgar al paciente un resultado funcional auditivo (aceptable dentro de los 25db), adaptar un audífono por vía aérea, o en aquellos pacientes con otorrea crónica y sospecha de colesteatoma por retención en un conducto muy estenótico.
Atresioplastía con fines funcionales:Los pacientes candidatos a este tipo de cirugía se definen según una escala de puntuación propuesta por Robert Jahrsdoerfer que establece puntajes según los siguientes parámetros tomográficos a fin de seleccionar los casos quirúrgicos con buen pronóstico funcional.
| • Estribo presente | 2 puntos |
| • Articulación incupoestapedial presente | 1 punto |
| • Complejo martillo-yunque | 1 punto |
| • Ventana oval normal | 1 punto |
| • Buen espacio del oído medio | 1 punto |
| • Buena neumatización mastoidea | 1 punto |
| • Trayecto nervio facial normal | 1 punto |
| • Ventana redonda presente | 1 punto |
| • Buen aspecto del pabellón (Microtia de 1° o 2°) | 1 punto |
Los autores utilizan la clasificación de Jahrsdoerfer de 10 puntos como factor pronóstico de la atresioplastía con tiempo funcional, pero, con una diferencia: se puntúa con 2 la presencia de articulación incudoestapedial, y no se tiene en cuenta el aspecto del pabellón.
Los mejores candidatos para la cirugía de atresioplastía tienen puntuaciones de 8 a 10 con un oído medio ventilado y vía ósea normal en la audiometría. Se recomienda un tratamiento alternativo para las puntuaciones por debajo de 6, audífono implantable por vía ósea.
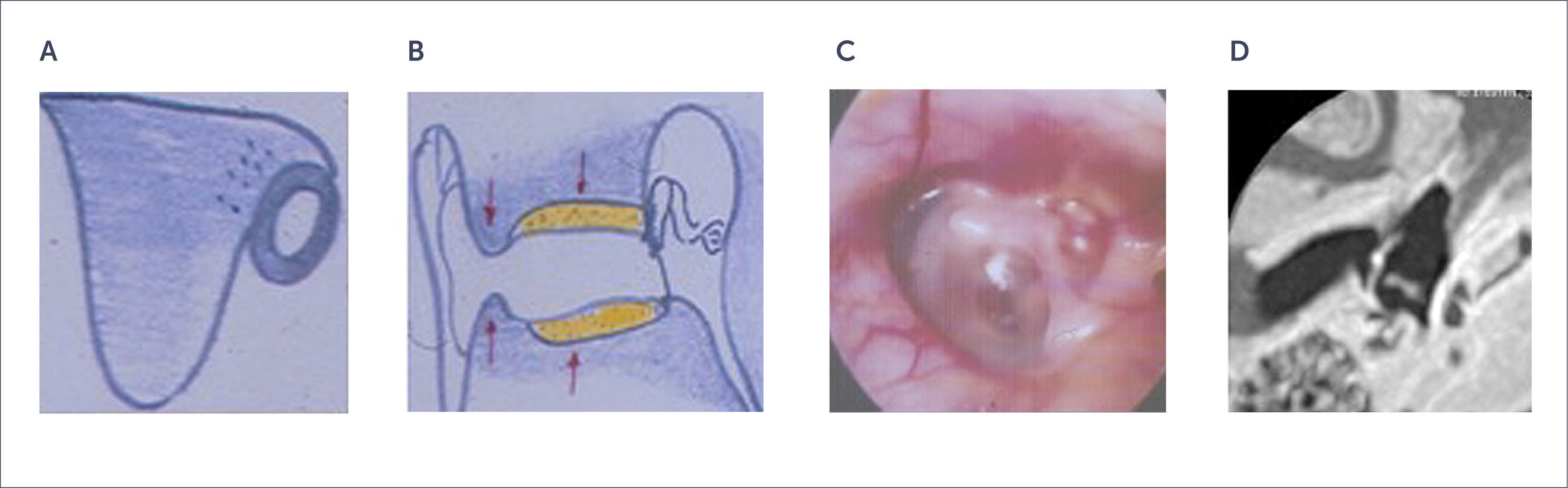
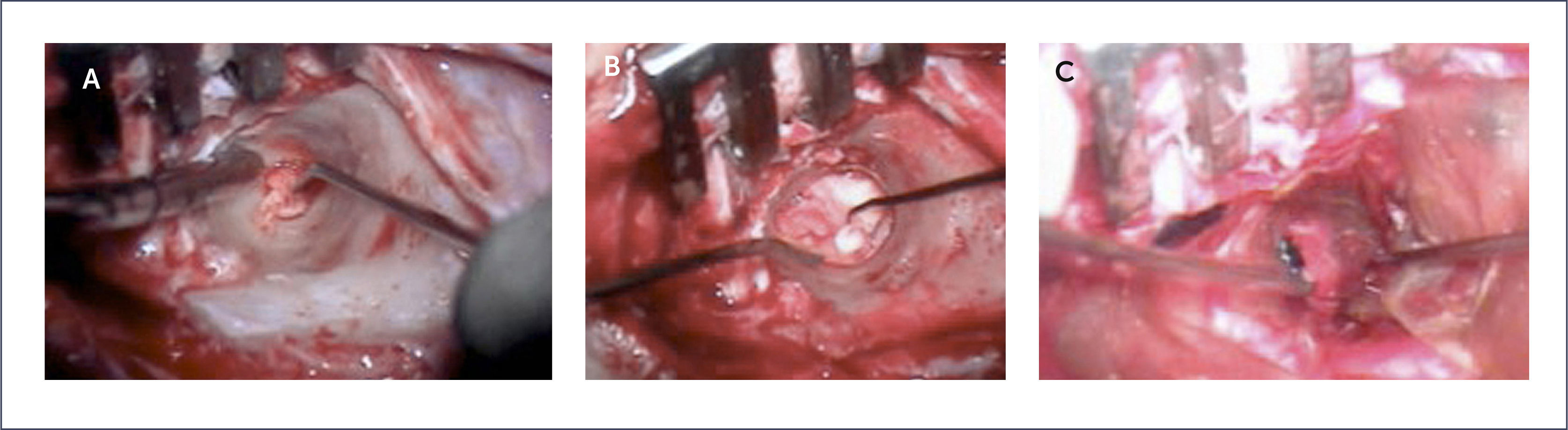
La técnica más aceptada en la actualidad es el abordaje anterógrado con amplia exposición de la caja timpánica y del epitímpano (Figura 9). La cirugía se realiza luego del tercer tiempo de la auriculoplastia (en caso de microtia de 3°) o como primer procedimiento en caso de microtias de 1° o 2°).
ATRESIOPLASTÍA PARA LA ADAPTACIÓN DE UN AUDÍFONO TRANSCANAL POR VÍA AÉREA Fresado de la placa atrésica formando el neoconducto; B) Condrotimpanoplastia; C) Tapizado con injerto de piel el neoconducto y el neotímpano.")
Esta alternativa se brinda al paciente cuando se realizó una atresioplastia con fines funcionales y los umbrales auditivos alcanzados no fueron satisfactorios para el paciente o cuando al realizar la puntuación por la escala de Jahrsdoerfer se comprueba que en la tomografía la cadena osicular se encuentra desarticulada.
Si la atresioplastía fue correctamente realizada, una vez epitelizado el neoconducto, se puede colocar el audífono transcanal por vía aérea, con buena tolerancia de las pieles injertadas (Figura 10).
OTRAS ALTERNATIVAS PARA LA HABILITACIÓN AUDITIVA
Para los pacientes que requieran una ayuda auditiva y en los cuales no sea viable la práctica de una atresioplastía o no sea la misma aceptada por el paciente o los padres.
Implantes de conducción ósea:B.A.H.A (Bone Anchored Hearing Aid)Los pacientes con disgenesia auditiva unilateral o bilateral que presenten hipoacusia conductiva o mixta con una vía ósea por encima de los 45dB, que no pueden emplear un audífono por vía aérea pueden ser candidatos al equipamiento con este tipo de dispositivos; por lo general se obtienen umbrales promedios entre 15 y 20 db de la vía ósea preoperatoria, con una ganancia funcional entre 35 y 40db.
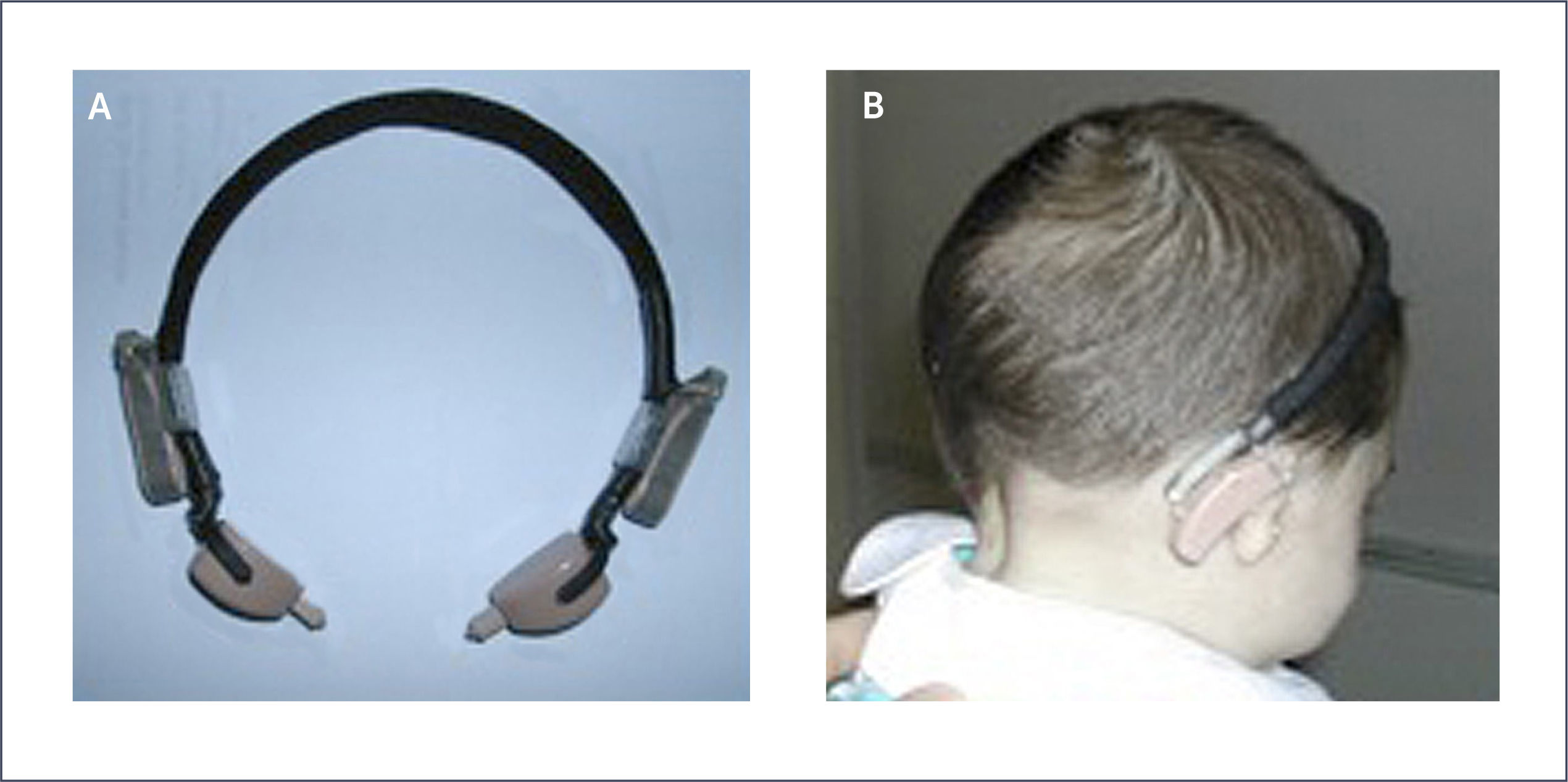
Este sistema permite realizar una prueba preoperatoria con una vincha o banda con un procesador de sonido mostrando al paciente el rendimiento auditivo. También se utiliza este tipo de vinchas en los niños pequeños, para su estimulación auditiva, ya que esta cirugía esta aprobada por la FDA para mayores de 5 años. (Figura 11 A y B).
 A) Vincha con dos procesadores de B.A.H.A.® B) Niño con microtia y vincha ósea.")
Existen dos opciones del sistema de BAHA:
BAHA Connect: consiste en un tornillo de titanio percutáneo, un empalme o conector y el procesador externo de sonido como lo descripto anteriormente.
BAHA Attract: consta de un tornillo de titanio que se implanta en el hueso, al igual que el BAHA Connect, se coloca luego un imán al mismo que se une y se oculta debajo de la piel (transcutáneo). El procesador de sonido se une a un imán externo y atrae al imán interno.
Bonebridge®Utiliza el sistema de conducción ósea estimulando a través de la vibración del cráneo la cóclea en forma directa. La parte que se implanta de este dispositivo consta de una bobina interna, un imán para mantener sujeto el procesador externo, un demodulador para convertir la señal desde el audio procesador y un Transductor de Masa Flotante de Conducción Ósea (BC-FMT) que genera las vibraciones del cráneo, la misma es de titanio y tiene una altura de 8.7mm y debe ser tallada una cazoleta en el cráneo para su anclaje.
Este sistema es transcutáneo y una vez cicatrizada la herida, se puede conectar el procesador externo.
Las indicaciones son las mismas que para el dispositivo BAHA.®
Se emplea en niños mayores de 5 años, con estudio tomográfico que permita evaluar que el grosor de la mastoides es compatible con la colocación del transductor de masa flotante.
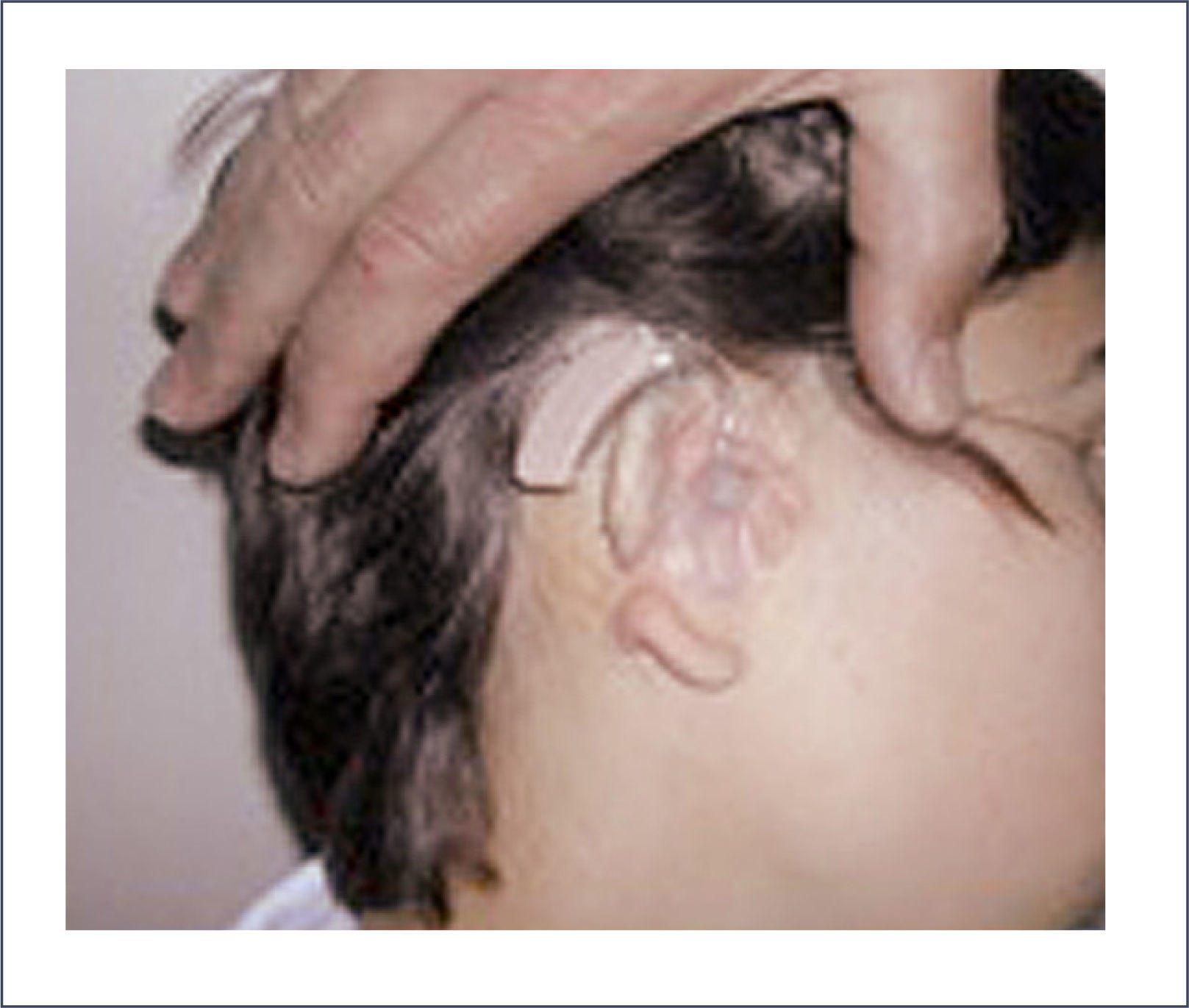
Sophono®Consiste en dos imanes fijados al cráneo por cinco tornillos de titanio, que no se osteointegran, en forma transcutánea. Se indica en niños mayores de 5 años (Figura 12 A y B).
Vibrant Soundbridge® Sophono® imanes sujeto al cráneo por 5 tornillos. B) Sophono conectado al procesador externo.")
Este dispositivo pertenece al grupo de implantes de oído medio que utilizan energía mecánica para estimular las estructuras del oído interno.
Diseñado para pacientes con hipoacusia conductiva, mixta, o sensorioneural de moderada a severa que, por razones médicas, no pueden usar un audífono, ya sea por disgenesia auditiva, o cuadros de dermatitis u otorrea que impiden la utilización de un audifono por vía aérea.
Aprobado para ser utilizado a partir de los 3 años de edad.
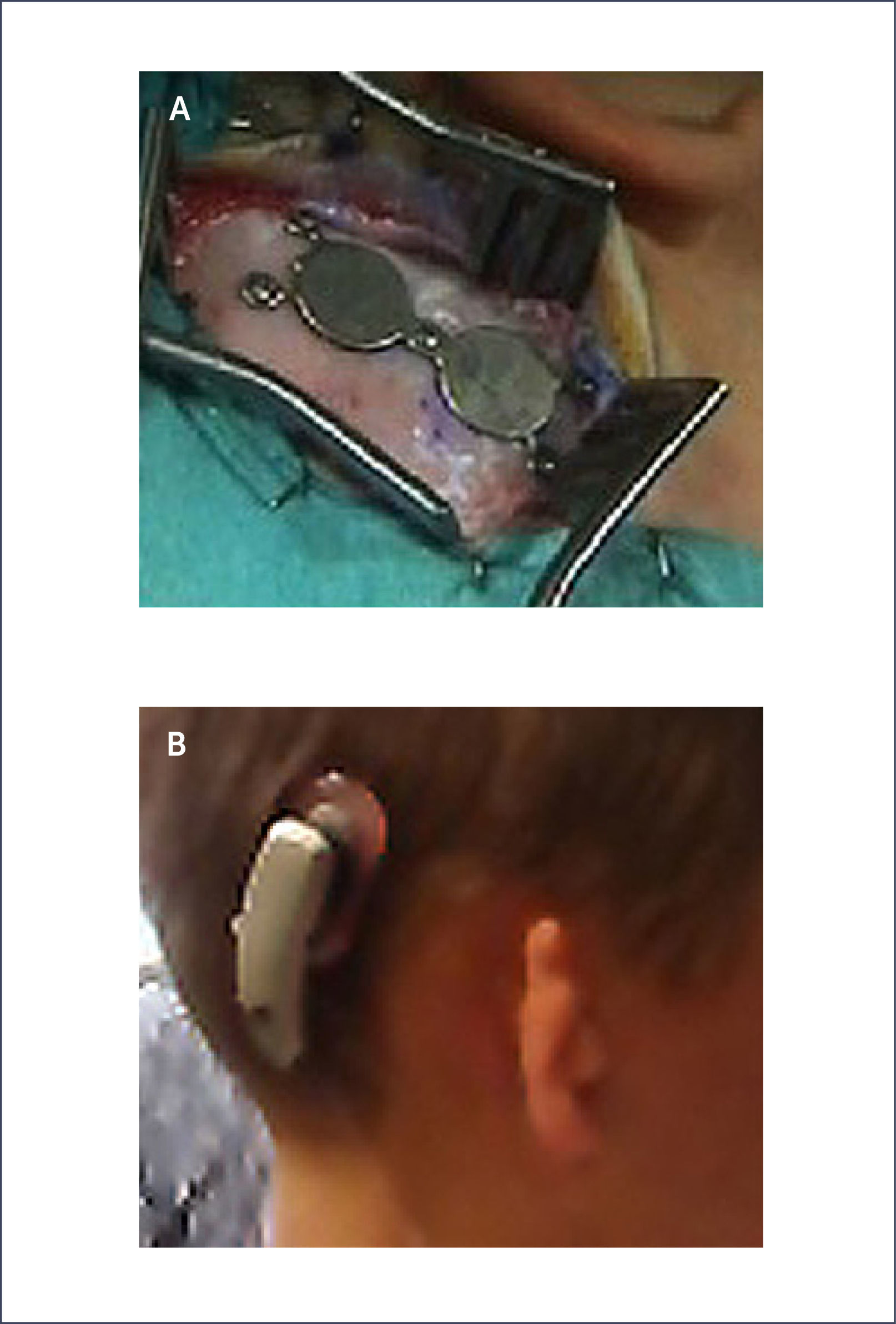
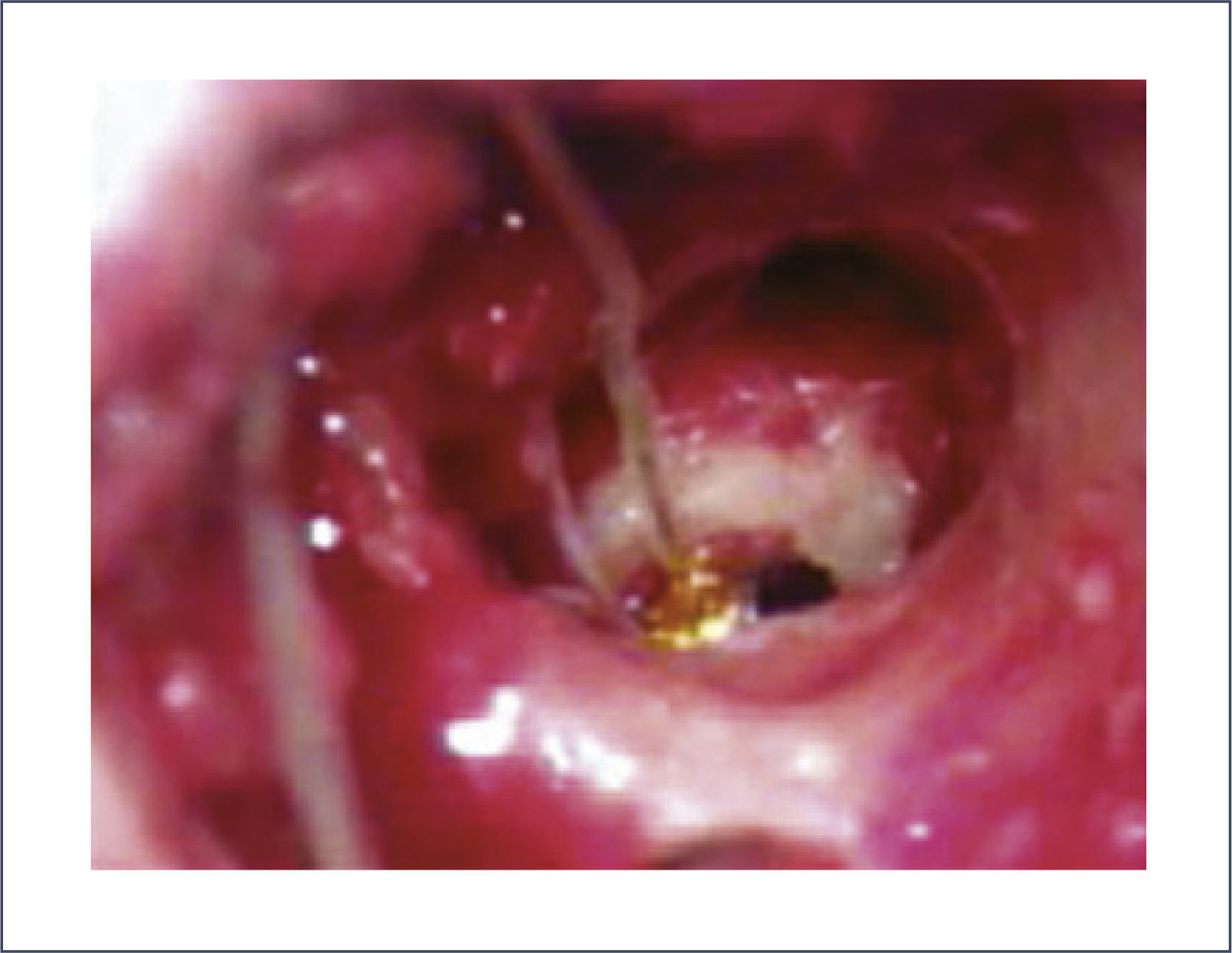
El procesador interno del Vibrant Soundbridge® es similar al empleado en los implantes cocleares, del mismo sale un electrodo de silicona que termina en un pequeño tambor cilíndrico (unidad vibrátil) de 1 x 2mm que produce una vibración electromagnética en su eje mayor. Este terminal puede ser colocado en varios sitios del oído medio: anclado sobre el yunque, el estribo, platina, o la ventana redonda (Figura 13).
EN SÍNTESIS
- 1.
La detención en el desarrollo embrionario por una determinada noxa generará un tipo de malformación, que será menos diferenciada a menor edad gestacional.
- 2.
El grado de disgenesia auditiva dependerá de la afectación del hueso temporal, del hueso timpanal y del pabellón auricular.
- 3.
Se debe sospechar colesteatoma por retención de queratina en las malformaciones con CAE muy estrecho, presenten o no otorrea.
- 4.
Los paciente con malformaciones del oído externo y/o medio presentan hipoacusia conductiva, generalmente no mayor a los 60db.
- 5.
Los pacientes que no presenten CAE permeable serán estudiados auditivamente mediante pruebas conductuales y potenciales evocados auditivos de tronco por vía ósea.
- 6.
En el tratamiento de las disgenesias auditivas se deben considerar dos aspectos: el estético reparativo del pabellón y el funcional auditivo, sobre todo si el caso es bilateral.
- 7.
Las disgenesias auditivas unilaterales con audición normal contralateral requerirán algún tipo de ayuda auditiva en caso de mal progreso en el lenguaje o bajo rendimiento escolar.
- 8.
Las disgenesias auditivas bilaterales deberán ser habilitadas auditivamente con otoamplífonos transcanal en caso de CAE permeable o mediante un sistema de conducción ósea en los primeros meses de vida.
Los autores declaran no tener conflictos de interés, en relación a este artículo.