


La hiperostosis cortical infantil, o enfermedad de Caffey-Silverman, es una entidad clínica caracterizada por neoformación ósea perióstica secundaria a un proceso inflamatorio agudo. De baja frecuencia, su curso clínico es generalmente autolimitado y de excelente pronóstico.
ObjetivoPresentar el caso de un lactante portador de un cuadro clínico compatible con una hiperostosis cortical infantil.
Caso clínicoLactante varón, 4 meses de edad, previamente sano, que consultó por presentar irritabilidad, llanto, aumento de volumen en la cara, los brazos y las piernas. Se observó aumento de volumen al nivel de la rama mandibular bilateral, simétrica, sensible, sin cambios en la coloración, la temperatura o la textura, hasta la región preauricular. El estudio bioquímico fue normal, y el estudio radiológico mostró reacción perióstica (periostitis e hiperostosis) al nivel de la rama mandibular, el fémur izquierdo, la tibia y el radio bilateral. Se manejó con antipiréticos, antiinflamatorios y analgésicos, y estuvo en observación en el servicio de urgencias durante 6h, donde se decidió su egreso y el manejo ambulatorio. La sintomatología cedió por completo entre 4 y 6 semanas después del alta.
ConclusiónLa hiperostosis cortical es una colagenopatía que debe ser considerada como diagnóstico diferencial en cuadros agudos de inflamación ósea, irritabilidad y fiebre. Es indispensable conocerla para sospecharla y la correlación clínico-radiológica es notable.
Infantile Cortical Hyperostosis, or Caffey-Silverman disease, is a rare condition characterised by generalised bone proliferation mediated by an acute inflammatory process. Diagnosis can be made through clinical evaluation and X-ray studies. The course is generally self-limiting and prognosis is excellent.
ObjectiveTo present the case of a 4-month child with clinical and radiological symptoms compatible with Infantile Cortical Hyperostosis.
Case reportA 4-month old male who presented with crying and irritability associated with swelling of the face, arms and legs was admitted to the Emergency Room of National Institute of Pediatrics. Bilateral mandibular swelling extending to periauricular region was observed, with no signs of inflammation. X-ray studies showed a periosteal reaction in the jaw, left femur and tibia, and radius bilateral. Clinical observation combined with analgesics and antipyretics was the only medical intervention. Four to six months after discharge from hospital, the symptoms disappeared, confirming the good prognosis of this condition.
ConclusionInfantile cortical hyperostosis is a collagenopathy, which must be considered as a differential diagnosis in acute bone inflammatory processes, irritability and fever. It is important to understand and identify this disease and clinical-radiological correlation is remarkable.
La hiperostosis cortical infantil o enfermedad de Caffey-Silverman (también llamado Síndrome de Caffey) es una entidad clínica caracterizada por neoformación ósea perióstica secundaria a un proceso inflamatorio1. Puede presentarse de manera prenatal o en los primeros años de vida, y generalmente es autolimitada. Se manifiesta con dolor/irritabilidad secundarios al aumento de volumen y engrosamiento del periostio, tejido blando y conectivo circundante en un área localizada. El episodio agudo generalmente se acompaña de fiebre y elevación sérica de marcadores proinflamatorios. La fiebre e irritabilidad en el paciente representan los principales motivos de consulta en los servicios de urgencia pediátrica, y en su etiología deben considerarse causas infecciosas y no infecciosas, como en este caso.
Esta enfermedad fue descrita por primera vez a mediados del siglo xx correlacionando la clínica con hallazgos radiológicos particulares2. Es una enfermedad rara, de la que no se ha determinado la incidencia; sin embargo, hay menos de 150 casos descritos desde su reconocimiento3. La mayor parte de los casos son hereditarios y el patrón de herencia para la mutación más estudiada (gen COL1A1, 17q21) es autosómico dominante con penetrancia incompleta4. En el caso presentado no se logró realizar estudio para determinar la presencia de mutación puntual. Recientemente se ha identificado una forma prenatal (muy grave/letal) cuyo patrón de herencia es probablemente autosómico recesivo1. La enfermedad ocurre en los primeros 6 meses de edad, con presentación inicial de sintomatología a las 9 semanas de vida5. No se ha descrito predilección por el género.
La historia natural de la enfermedad ha demostrado que los casos son autolimitados6. Se presenta como un «ataque masivo»7 de neoformación subperióstica. Hay engrosamiento localizado de la corteza ósea e inflamación del tejido blando circundante. El hueso que se afecta con mayor frecuencia en la forma esporádica es la mandíbula, aunque las lesiones en la parrilla costal y las clavículas son muy frecuentes8. En la forma hereditaria se describe mayor afección en la tibia9. Radiológicamente las corticales del hueso afectado presentan ensanchamiento y un borde irregular y adelgazado10. Tras la resolución de la sintomatología hay baja recurrencia y las secuelas del proceso inflamatorio reportadas con escasas.
El diagnóstico diferencial debe realizarse con otras enfermedades que cursen con inflamación ósea, como osteomielitis, hipervitaminosis, escorbuto, tumores óseos y síndrome de niño maltratado (variedad abuso físico), así como procesos infecciosos en todos los casos.
El objetivo de este trabajo es presentar un caso de una colagenopatía poco frecuente, que se benefició de una sospecha clínica temprana con correlación radiológica.
Caso clínicoLactante varón de 4 meses de edad, proveniente del Estado de México, previamente sano, producto de la tercera gesta de una madre de 40 años. Nació de término, con peso adecuado para la edad gestacional. Hitos de desarrollo y parámetros de crecimiento adecuados para edad. Ingresó en el servicio se urgencias por presentar un cuadro de 20 días de evolución caracterizado por irritabilidad y llanto, aumento de volumen en la cara, los brazos y las piernas. A su ingreso se constató una adecuada coloración mucocutánea, sin dificultad respiratoria. La madre no refirió historia de fiebre, viajes o cuadros infecciosos recientes, reportando una adecuada ingesta, diuresis y evacuaciones sin alteraciones. Vacunación completa de acuerdo a edad. No existía consanguinidad o endogamia, como tampoco antecedentes de traumatismo.
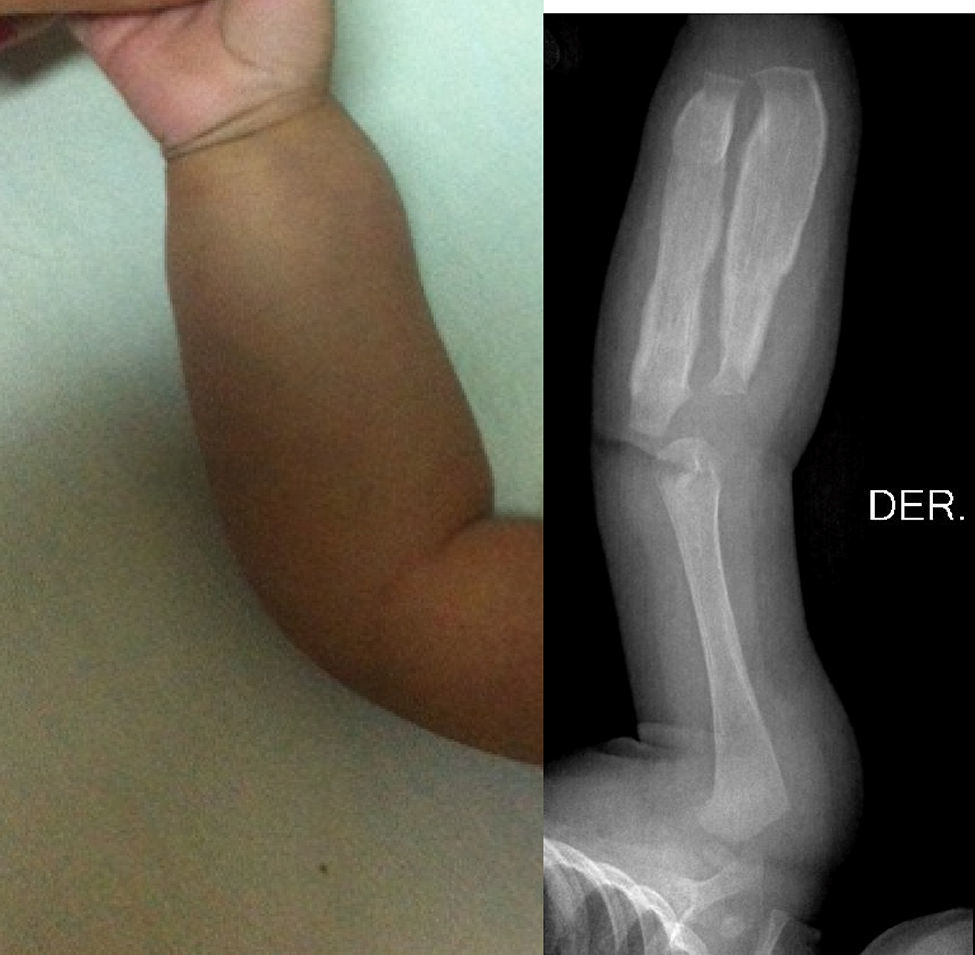
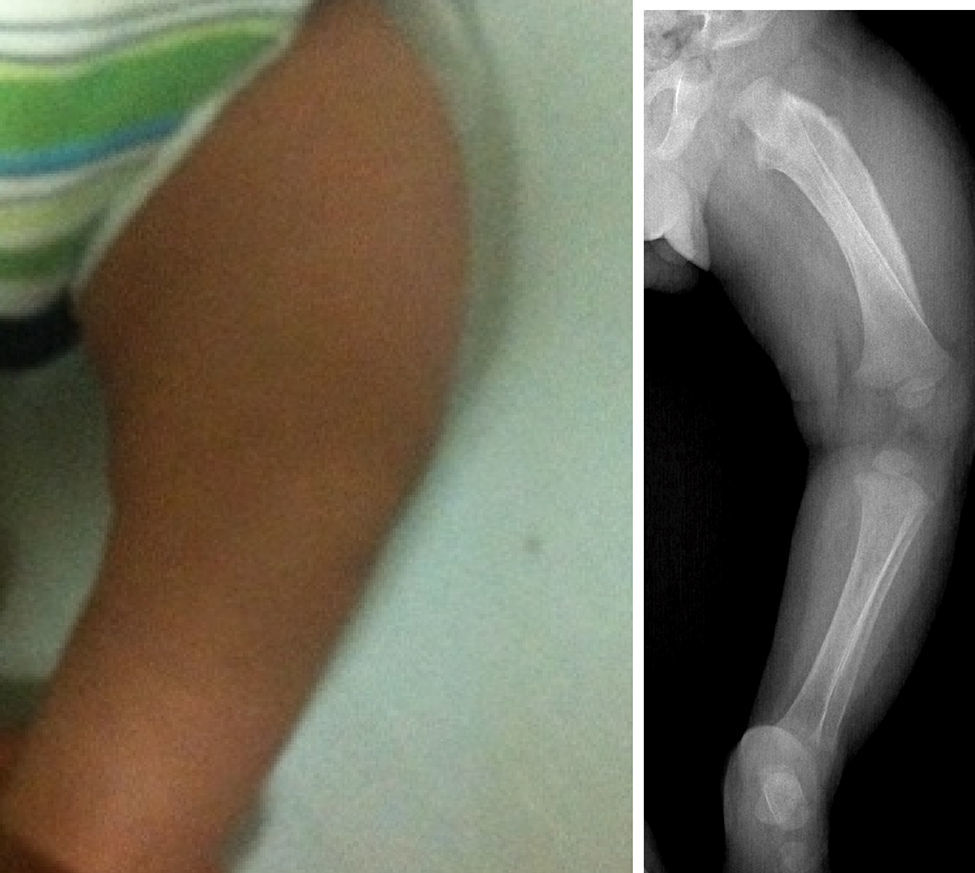
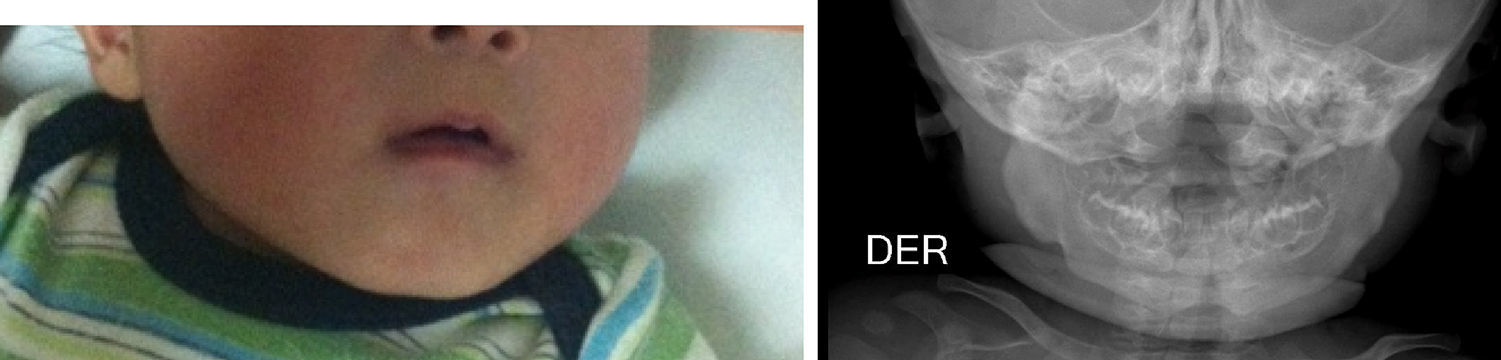
A la exploración física el niño se encontraba muy irritable, con dolor, llanto a la manipulación y al movimiento pasivo en miembros torácicos y pélvico izquierdo. Hemodinámicamente estable, con taquicardia leve asociada al llanto. No se apreciaban dismorfias, adecuado estado nutricional, bien hidratado, con limitación discreta en los movimientos de brazos y piernas. Presentó fiebre de bajo grado, sin evidencias de respuesta inflamatoria sistémica ni de hipoperfusión. En el cráneo se observó un aumento de volumen al nivel de la rama mandibular bilateral, simétrica, sin cambios en la coloración, temperatura o textura de la piel; llanto al tacto del borde mandibular, sin crepitación a la palpación. Se constató aumento de volumen hasta región preauricular. No se palparon masas ni adenomegalias. Se evidenció un aumento de volumen en los antebrazos y en el muslo izquierdo, con eritema leve, doloroso. El resto de la exploración física fue normal. No se observó equimosis ni datos de sangrado o heridas.
Los exámenes de laboratorio reportaron en la citometría hemática anemia leve con Hb 11g/dl y Hto 33%, normocítica normocrómica. Leucocitosis a expensas de neutrofilia (LT 20.400/mm3, N88%), sin bandas. Plaquetas en 384.000. Velocidad de sedimentación 55mm/seg, proteína C reactiva 3mg/dl. Elevación de fosfatasa alcalina y deshidrogenasa láctica. Electrolitos séricos normales con fósforo en 4mEq/l y calcio en 8,9mEq/l. El examen general de orina no presentaba parámetros patológicos. Los cultivos de sangre y orina no presentaron desarrollo bacteriano.
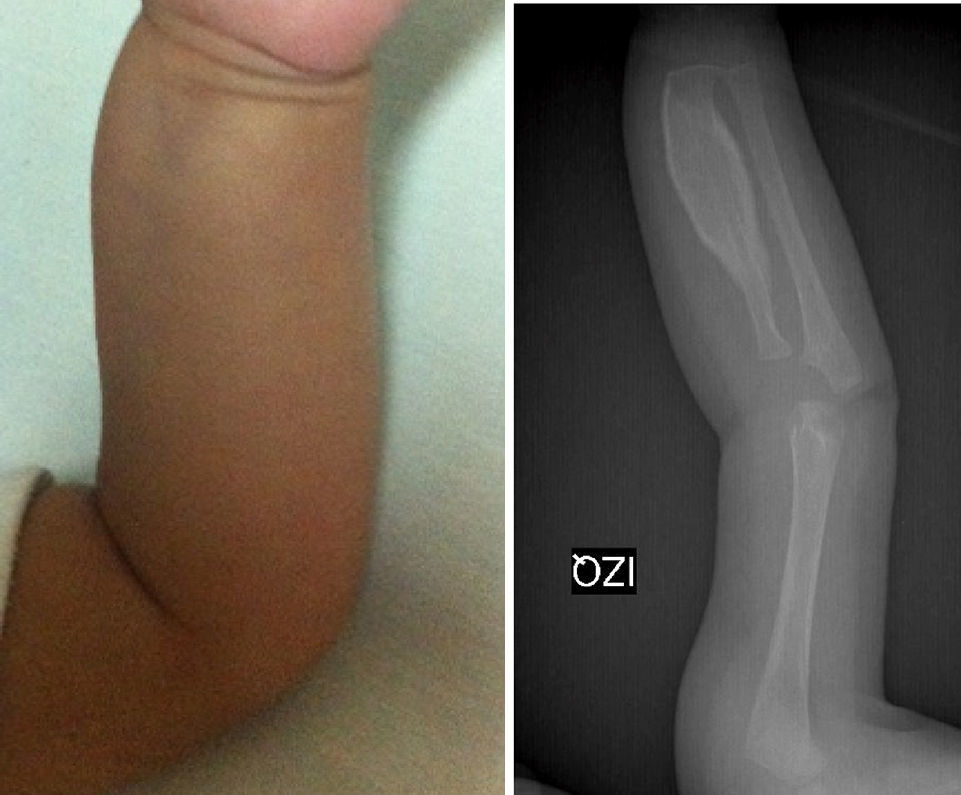
Los estudios de imágenes solicitados fueron radiografías simples de diferentes segmentos corporales, que demostraron una reacción perióstica (periostitis e hiperostosis) con cambios escleróticos, sin lesiones osteolíticas. Se apreció compromiso de la rama mandibular, el fémur izquierdo, la tibia y el radio bilateral. No se observaron microfracturas ni lesiones que involucraren metáfisis (figs. 1–4).



Pierna izquierda.
Estudio radiográfico de pierna izquierda. Proyección AP que demuestra afección severa en el fémur izquierdo. Nótese hiperostosis discreta en la cara anterior de la tibia izquierda y el engrosamiento cortical del radio. Los núcleos de osificación y las epífisis no presentan cambios.

Se realizó el diagnóstico de forma clínica, y se inició manejo antipirético, antiinflamatorio y analgésico; se vigiló al paciente en el servicio de urgencias durante 6h, y se decidió su egreso y manejo ambulatorio. La sintomatología cedió por completo entre 4 y 6 semanas después del alta.
DiscusiónNo se conoce de manera clara la fisiopatología en la hiperostosis cortical infantil; recientemente se ha propuesto el papel que representa el colágeno tipo i, por lo que debe considerarse como una colagenopatía de curso autolimitado y benigno. Se ha localizado una mutación puntual con sustitución de arginina por cisteína en la cadena alfa uno del colágeno tipo i, aunque no se ha logrado definir el enlace funcional entre dicha mutación y los mecanismos fisiopatológicos que relacione la alteración estructural del colágeno, que desencadena una cascada proinflamatoria autolimitada.
Debe sospecharse y reconocerse clínicamente para evitar procedimientos invasivos para su diagnóstico11. El método de imágenes indicado es la radiografía simple del segmento corporal afectado. Se han descrito hallazgos en otros estudios de imagen como tomografía axial computarizada, resonancia magnética, así como resultados histopatológicos donde solo se evidencia inflamación, por lo que no está recomendado su realización en todos los casos. El diagnóstico es clínico-radiológico.
La enfermedad se caracteriza por neoformación perióstica y engrosamiento cortical que produce inflamación en el tejido muscular y conectivo circundantes. Se afecta la porción diafisiaria en los huesos tubulares y respeta la epífisis y la metáfisis. Puede afectar cualquier hueso, con excepción de las falanges, los cuerpos vertebrales y los huesos cuboideos12. No afecta la fisis de crecimiento13. Cuando la reacción subperióstica es muy importante entre huesos paralelos, se genera un aumento de presión y pérdida del tejido perióstico, con lo que puede ocurrir una fusión entre ambas corticales, descrito como puente interóseo. Esto puede resultar en secuelas y deformaciones óseas14; se han descrito casos de formas recurrentes15. Sin embargo, lo más frecuente es que la reacción perióstica se autolimite, se forme nuevo hueso subperióstico y el excedente de hueso periférico sea eliminado por remodelación16.
Los hallazgos radiológicos son la hiperostosis subperióstica cortical (engrosamiento perióstico) principalmente. Existe neoformación ósea subperióstica con incremento en densidad del grosor de la corteza17.
Los cambios histopatológicos tempranos demuestran lesiones confinadas al periostio. Se ha indagado en la relación entre alteraciones estructurales puntuales del colágeno tipo i y la respuesta proinflamatoria, y se ha propuesto que posterior a la mutación puntual (en el caso de la forma AD) hay dominios en la cadena mutada del colágeno que interactúan tanto con la interleuquina 218 como con la fibronectina19,20, asimismo se establece bien el papel de PGE y COX2 y TGFbeta21 como mediadores proinflamatorios importantes.
El tratamiento consiste en revertir el proceso inflamatorio. Se ha reportado resolución de la sintomatología con antiinflamatorios no esteroideos (específicamente naproxeno e indometacina); recientemente se ha propuesto la eficacia del manejo con antiinflamatorios esteroideos como segunda línea terapéutica, ya que se propone que aceleran la remodelación ósea.
El pronóstico es generalmente favorable, el cuadro remite sin administración de medicamentos entre los 6 y 9 meses. En pocos casos se ha reportado recurrencia o secuelas y las primeras se presentan en la adolescencia temprana. Generalmente se presenta con regresión espontánea de la sintomatología. Las hiperostosis remiten en los primeros 12 meses22. El diagnóstico puede realizarse fácilmente por los médicos de primer contacto (correlación clínica y radiológica), evitando métodos diagnósticos excesivos e invasivos.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores han obtenido el consentimiento informado de los pacientes y/o sujetos referidos en el artículo. Este documento obra en poder del autor de correspondencia.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
