





La tomografía por emisión de positrones (PET) es una técnica de imagen que en los últimos años ha experimentado un gran crecimiento y se considera una herramienta básica de gran utilidad en el campo de la oncología, sin olvidar sus indicaciones en otras áreas como la neurología y la cardiología. Aunque la 18F-fluorodeoxyglucose (18F-FDG) es el radiofármaco más ampliamente utilizado en PET, disponer de nuevos radiotrazadores ha sido un elemento clave en la expansión de esta técnica porque han permitido estudiar diferentes dianas biológicas esenciales para un mayor conocimiento y una mejor caracterización de diferentes patologías, contribuyendo de esta forma a la investigación y el desarrollo de nuevos agentes terapéuticos. En este trabajo se hace una descripción de varios radiofármacos para PET, estructurada según su área de aplicación. Algunos de ellos ya están disponibles comercialmente, mientras que otros todavía están en fase de investigación o pendientes de autorización.
Positron emission tomography (PET) is an imaging technique that has grown greatly in recent years. PET is considered a fundamental tool in oncology, and it also has indications in other fields such as neurology and cardiology. Although 18F-fluorodeoxyglucose (18F-FDG) is the radiopharmaceutical most widely used in PET, the availability of new radiotracers has been a key element in the expansion of the use of PET. These new radiopharmaceuticals have made it possible to study different biological targets that are essential for obtaining greater knowledge and better characterization of different diseases and have thus contributed to the research and development of different therapeutic agents. This article provides a description of different PET radiopharmaceutical, structured according to their areas of application. Some of these radiotracers are already commercially available, whereas others are still under research or pending approval by regulatory bodies.
Los radiofármacos constituyen un tipo de medicamentos especiales que se definen como cualquier producto que, cuando está preparado para su uso con finalidad terapéutica o diagnóstica, contiene uno o más radionúclidos1. Debido a su carácter radiactivo y a la necesidad, en la mayoría de los casos, de someterlos a un proceso de preparación antes de su uso, es muy importante establecer una buena coordinación entre las unidades de radiofarmacia y los servicios de medicina nuclear para gestionar y programar de forma óptima las diferentes exploraciones diagnósticas o procedimientos terapéuticos solicitados.
Los radiofármacos son medicamentos radiactivos constituidos por un radionúclido que emite un determinado tipo de radiación y un vehículo que proporciona al radiofármaco la capacidad de dirigirse hacia un órgano o tejido diana, por el que debe presentar un alto grado de especificidad y selectividad2.
Las posibles transformaciones que sufren los núcleos radiactivos en su proceso de estabilización dan lugar a distintos tipos de emisiones que determinarán su uso. Así, aquellos radiofármacos constituidos por radionúclidos emisores de partículas α (núcleos de helio) o β− (electrones) están indicados en terapia, mientras que los que contienen emisores de β+ (positrones) o de radiación γ se administran para la realización de exploraciones diagnósticas3. Los radiofármacos utilizados en la tomografía por emisión de positrones (PET) son considerados agentes de imagen molecular, ya que permiten la visualización, la caracterización y la medida de procesos biológicos celulares y moleculares en un organismo vivo de forma no invasiva4. Por ello, es posible la detección temprana de procesos patológicos ya que, generalmente, los cambios biomoleculares anteceden a los cambios anatómicos5. En el caso concreto de los pacientes oncológicos, la imagen de PET revela aspectos de la bioquímica subyacente en el tumor antes y durante el tratamiento, lo que contribuye a individualizar la atención de este tipo de pacientes6.
Los radiofármacos para PET contienen radionúclidos emisores de positrones. Estas partículas se producen durante la desintegración radiactiva de un núcleo con exceso de protones respecto al número de neutrones, de modo que un protón se transforma en un neutrón, emitiendo simultáneamente un positrón y un neutrino (que no interviene en la formación de la imagen). El positrón (electrón positivo o antielectrón) va perdiendo energía tras sucesivas colisiones y cuando está prácticamente en reposo se aniquila con un electrón del medio, de forma que las masas de ambos se convierten en energía y, en concreto, se producen dos fotones de 511KeV cada uno, que son emitidos de manera simultánea en sentidos opuestos y son detectados por el tomógrafo (detección por coincidencia). Así, es posible la identificación de la línea a lo largo de la cual se emitieron y, por tanto, una mejor localización de la acumulación del radiofármaco para la formación de imágenes.
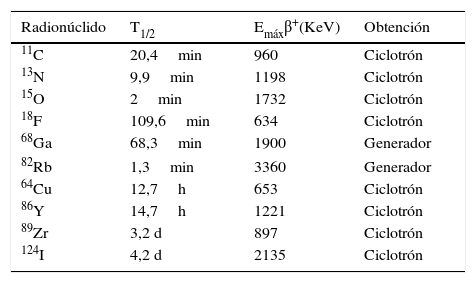
En la tabla 1 se muestran algunos radionúclidos para PET con sus correspondientes periodos de semidesintegración (T1/2), energías máximas del positrón (Emáxβ+) y métodos de obtención (ciclotrón/generador). Los más utilizados para la síntesis de radiofármacos PET son los situados en la primera mitad de la tabla. Se caracterizan por tener, la mayoría de ellos, un T1/2 muy corto, lo que limita de forma importante su disponibilidad. El T1/2 influye no solo en la síntesis y la distribución del radiofármaco, sino que también es importante que sea compatible con el proceso biológico en estudio, su cinética y su aclaramiento sanguíneo6. Por tanto, los radionúclidos con un T1/2 más largo (cobre-64, itrio-86, zirconio-89, iodo-124) permiten desarrollar radiofármacos basados en moléculas que presenten una farmacocinética lenta, como es el caso de los anticuerpos que requieren 2-4 días para obtener imágenes con un contraste óptimo para su análisis. La mayoría de los radionúclidos PET se producen en ciclotrón, excepto el galio-68 (68Ga) y el rubidio-82 (82Rb), que se obtienen a partir de un generador. La elección de un determinado radionúclido depende, fundamentalmente, de su disponibilidad y de sus características fisicoquímicas7.
Principales radionúclidos para tomografía por emisión de positrones
| Radionúclido | T1/2 | Emáxβ+(KeV) | Obtención |
|---|---|---|---|
| 11C | 20,4min | 960 | Ciclotrón |
| 13N | 9,9min | 1198 | Ciclotrón |
| 15O | 2min | 1732 | Ciclotrón |
| 18F | 109,6min | 634 | Ciclotrón |
| 68Ga | 68,3min | 1900 | Generador |
| 82Rb | 1,3min | 3360 | Generador |
| 64Cu | 12,7h | 653 | Ciclotrón |
| 86Y | 14,7h | 1221 | Ciclotrón |
| 89Zr | 3,2 d | 897 | Ciclotrón |
| 124I | 4,2 d | 2135 | Ciclotrón |
Emáxβ+: energía máxima del positrón; T1/2: periodo de semidesintegración.
Los radionúclidos orgánicos, carbono-11 (11C), nitrógeno-13 (13N) y oxígeno-15 (15O), son isótopos de elementos presentes en las biomoléculas, por lo que los radiofármacos correspondientes tendrán el mismo comportamiento biológico que la molécula sin marcar. Sin embargo, esta característica hace que el radiofármaco, al ser indistinguible por el organismo, siga la misma ruta metabólica que su análogo sin marcar, y por tanto la medida de su distribución en los tejidos requiere un análisis matemático complejo2,6,8.
Entre los radionúclidos del grupo de los halógenos, el flúor-18 (18F) es el más utilizado ya que, aun no siendo un elemento habitual en moléculas biológicas, presenta una serie de propiedades que hacen de él el radionúclido ideal para la síntesis de radiofármacos PET. El radio atómico del flúor es muy similar al del hidrógeno, por lo que puede esperarse que, en una molécula orgánica determinada, el enlace carbono-flúor simule el comportamiento biológico de la unión carbono-hidrógeno. Por otra parte, la electronegatividad del flúor es muy distinta de la del hidrógeno, lo que afecta a las propiedades fisicoquímicas de la molécula8. Por tanto, no puede asumirse que una molécula y su análogo fluorado tengan el mismo comportamiento en cuanto a biodistribución, unión a proteínas, afinidad por receptores o transportadores, y metabolización. Sin embargo, estas modificaciones pueden ser beneficiosas para usar el análogo fluorado como radiofármaco, pues al sustituir un átomo de hidrógeno por uno de flúor se obtiene un compuesto que se comporta como un antimetabolito, sufre atrapamiento metabólico y, por tanto, presentará una farmacocinética que puede asimilarse a un modelo matemático sencillo que permita su cuantificación. En cuanto al isótopo radiactivo, el 18F presenta también una serie de ventajas: se produce en gran cantidad en ciclotrón; su energía de emisión (634KeV), menor que la de otros isótopos PET, permite obtener imágenes de mejor resolución; además, contribuye a que la dosis absorbida por el paciente sea menor y, sobre todo, su T1/2 de 110 minutos facilita la síntesis de radiofármacos más o menos complejos y permite la distribución de los radiofármacos fluorados a centros satélites relativamente alejados del ciclotrón, aparte de ofrecer la posibilidad de realizar estudios de varias horas4,8,9.
El 68Ga es el radionúclido del grupo de los elementos metálicos con mayor proyección en el campo de la radiofarmacia PET, debido a su disponibilidad a partir del generador germanio-68/galio-68. Los T1/2 del padre 68Ge (270 días) y del hijo 68Ga (68min) hacen que la vida útil del generador sea de aproximadamente 1 año y que puedan realizarse síntesis de radiofármacos más o menos complejas7. A diferencia del 18F y del 11C, que forman enlaces covalentes con la molécula que actúa de vehículo, la química del 68Ga se basa en la formación de compuestos de coordinación a través de agentes quelantes bifuncionales, siendo especialmente útil para el marcaje de péptidos, proteínas y fragmentos de anticuerpos u otro tipo de pequeñas moléculas, tipo affibodies y nanobodies, que presenten un rápido aclaramiento sanguíneo y con alta afinidad por la diana seleccionada10.
El creciente interés por el 68Gaen los últimos años se debe, fundamentalmente, al desarrollo de nuevos generadores cuyos eluidos facilitan la síntesis del radiofármaco, así como de nuevos péptidos que pueden ser marcados y de un gran número de quelantes que permiten formar enlaces estables7,11.
El vehículo utilizado en la síntesis de un determinado radiofármaco dependerá del proceso a evaluar. Es el que confiere las características biológicas y las interacciones químicas y bioquímicas del radiofármaco en el interior del organismo. Debe presentar una afinidad selectiva por la diana hacia la que va dirigida. Estas dianas (targets) pueden ser receptores, transportadores, antígenos o situaciones provocadas por una patología, como alteraciones metabólicas específicas, situaciones de hipoxia, cambios en la vascularización, etc.2.
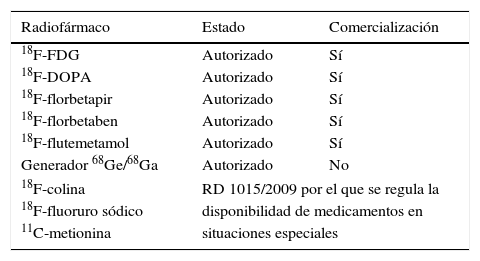
Durante los últimos años, los radiofármacos PET han demostrado su utilidad como agentes de imagen molecular principalmente en tres áreas clínicas: oncología, neurología y cardiología; en la primera es en la que más aplicaciones se han desarrollado. A pesar de ello, salvo la 18F-fluorodesoxiglucosa (18F-FDG), muy pocos radiotrazadores PET son de uso sistemático, ya que su disponibilidad está relacionada con sus condiciones de producción y logística de distribución, claramente dependientes de las características físicas y del método de obtención (ciclotrón o generador) del radionúclido presente en la molécula. Además, hay que considerar que los radiofármacos PET, como medicamentos que son, deben someterse a un procedimiento de registro y autorización por parte del organismo competente de cada país, que puede ser largo y costoso, y que retrasa la incorporación de nuevos radiofármacos por los canales habituales de comercialización. En España es la Agencia Española de Medicamentos y Productos Sanitarios (AEMPS) la que realiza la evaluación de un radiofármaco previa a su autorización para uso clínico, uso compasivo o uso en ensayos clínicos, conforme a la Ley 29/2006, de 26 de julio, de garantías y uso racional de los medicamentos y productos sanitarios. Actualmente, los radiofármacos PET autorizados por la AEMPS son los siguientes: 18F-FDG, 18F-FDOPA, 18F-florbetapir, 18F-florbetaben, 18F-flutemetamol y generador de 68Ge/68Ga, aunque este último aún no está comercializado. Existen otros radiofármacos PET, como la 18F-colina, el 18F-fluoruro sódico y la 11C-metionina que están disponibles por la vía del Real Decreto 1015/2009, por el que se regula la disponibilidad de medicamentos en situaciones especiales (tabla 2).
Situación administrativa en España de los radiofármacos para tomografía por emisión de positrones
| Radiofármaco | Estado | Comercialización |
|---|---|---|
| 18F-FDG | Autorizado | Sí |
| 18F-DOPA | Autorizado | Sí |
| 18F-florbetapir | Autorizado | Sí |
| 18F-florbetaben | Autorizado | Sí |
| 18F-flutemetamol | Autorizado | Sí |
| Generador 68Ge/68Ga | Autorizado | No |
| 18F-colina | RD 1015/2009 por el que se regula la | |
| 18F-fluoruro sódico | disponibilidad de medicamentos en | |
| 11C-metionina | situaciones especiales | |
Fuente: http://www.aemps.gob.es/
La introducción de la PET-TC ha contribuido de manera significativa al mayor conocimiento y la mejor caracterización de diferentes tumores. Se ha convertido en una herramienta fundamental en la atención del paciente oncológico y desempeña un papel muy importante en las diferentes etapas de la enfermedad, destacando su utilidad en la detección temprana y su influencia en la selección del tratamiento.
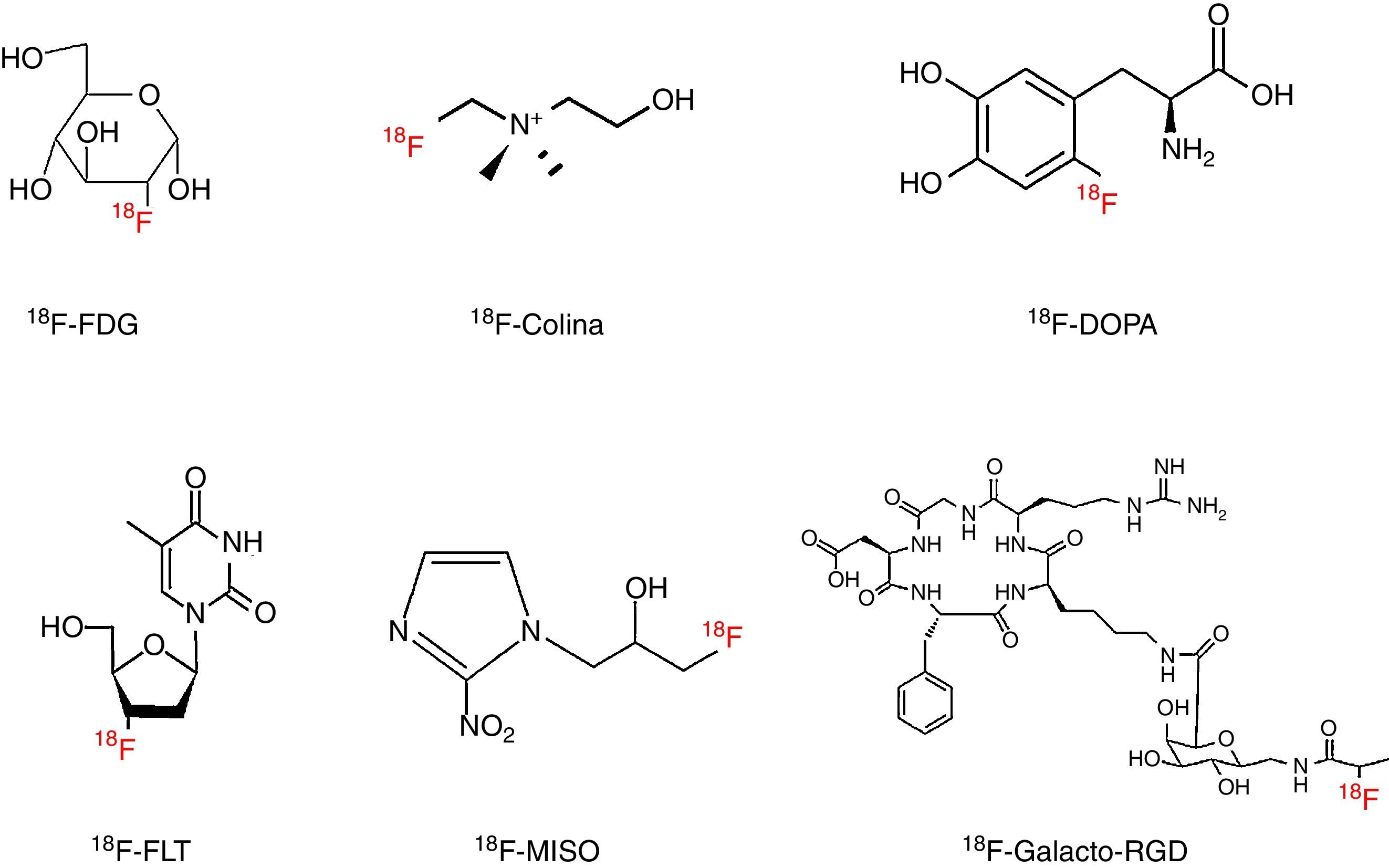
18F-FDG: 2-[18F]fluoro-2-desoxi-D-glucosaLa 18F-FDG es el radiofármaco PET más utilizado en la práctica clínica diaria. Se trata de un análogo de la glucosa que contiene un átomo de 18F en el carbono 2 en lugar del grupo hidroxilo de la molécula original (fig. 1). Su uso en oncología se basa en el mayor consumo de glucosa por parte de las células tumorales, debido fundamentalmente a un aumento de los transportadores de glucosa y a una mayor actividad de la hexocinasa6. Es un marcador del metabolismo glucídico, de modo que la concentración de 18F-FDG refleja el grado de metabolismo necesario para mantener una alta tasa de crecimiento o proliferación, y por tanto proporciona una medida indirecta de la proliferación celular12.

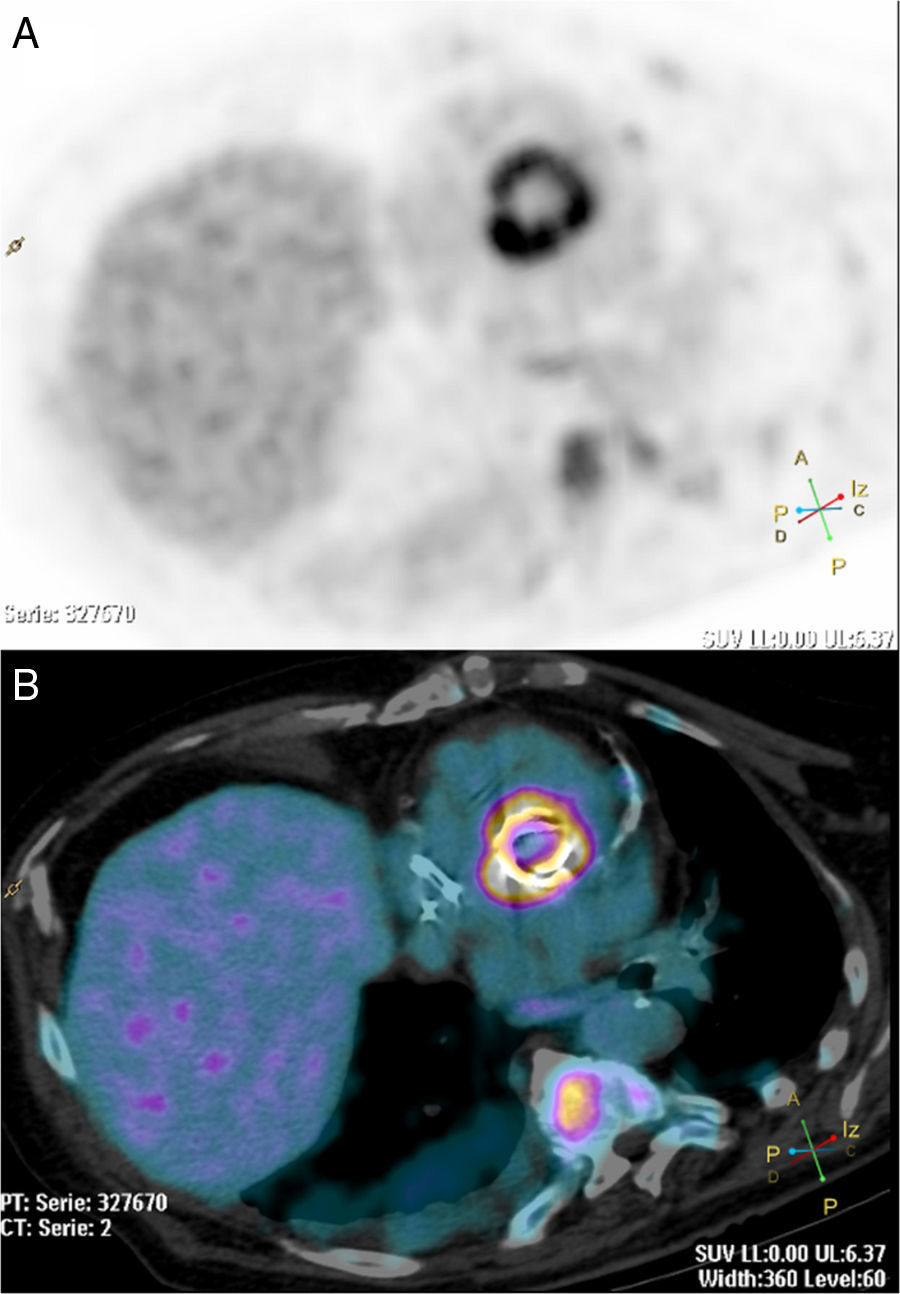
Sus principales indicaciones son el diagnóstico, la estadificación (fig. 2) y la reestadificación, en especial en pacientes asintomáticos con elevación de los marcadores tumorales, así como la monitorización de la respuesta al tratamiento, permitiendo la optimización del régimen terapéutico de forma precoz.
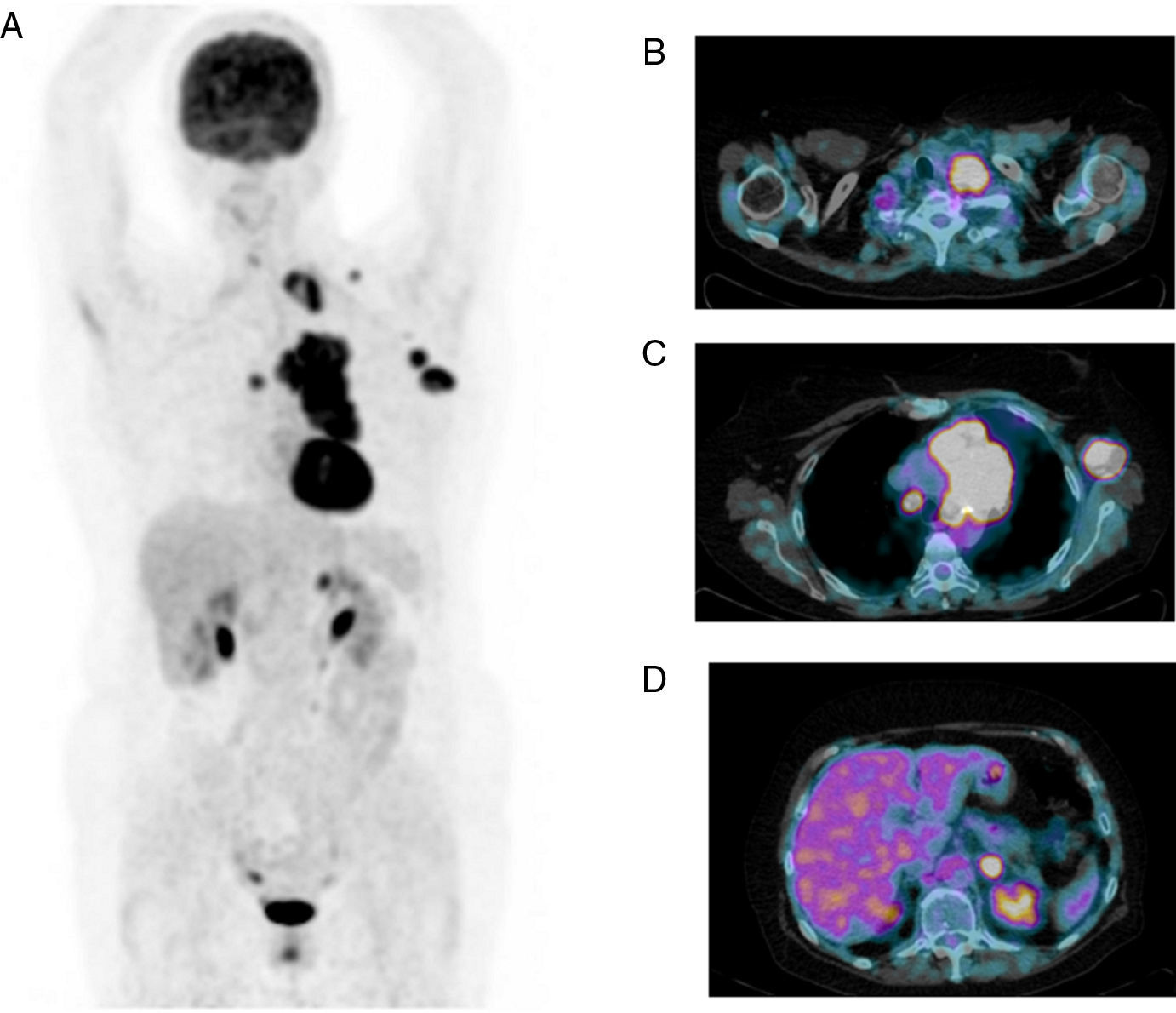
 (A) e imágenes de fusión PET-TC (B, C y D). Mujer de 62 años de edad remitida para estadificación de neoplasia de pulmón (T4N3M1b). Se observa una masa hipermetabólica polilobulada hiliar izquierda con infiltración de la pleura mediastínica. Múltiples adenopatías hipermetabólicas mediastínicas bilaterales, hiliar izquierda y supraclavicular. Metástasis ganglionares (axilares izquierdas) y suprarrenal izquierda.")
Tomografía por emisión de positrones PET-TC con 18F-FDG. Imagen MIP (proyección de máxima intensidad) (A) e imágenes de fusión PET-TC (B, C y D). Mujer de 62 años de edad remitida para estadificación de neoplasia de pulmón (T4N3M1b). Se observa una masa hipermetabólica polilobulada hiliar izquierda con infiltración de la pleura mediastínica. Múltiples adenopatías hipermetabólicas mediastínicas bilaterales, hiliar izquierda y supraclavicular. Metástasis ganglionares (axilares izquierdas) y suprarrenal izquierda.
La 18F-FDG entra en la célula a través de los transportadores de glucosa e inicia la vía glucolítica con la fosforilación del carbono 6 por la hexocinasa. La siguiente etapa es la isomerización a fructosa 6 fosfato, para lo cual es imprescindible la intervención del grupo hidroxilo en el carbono 2 presente en la molécula de glucosa, por lo que la 18F-FDG, al carecer de este grupo funcional, no es sustrato de la isomerasa y, por tanto, sufre atrapamiento metabólico, es decir, el resultado final es la acumulación progresiva de la 18F-FDG en el interior de la célula4,6,13.
A pesar de que la PET con 18F-FDG es muy útil en el paciente oncológico, hay que reconocer que presenta una serie de limitaciones que hacen que su sensibilidad y su especificidad no sean óptimas en todos los tumores14. No es un marcador específico de malignidad, ya que también se acumula en zonas de infección o inflamación. Además, su captación fisiológica en el cerebro, los riñones y la vejiga dificulta el estudio de tumores localizados en esas zonas. En las neoplasias de crecimiento lento o altamente diferenciadas (cáncer de próstata, tumores neuroendocrinos [TNE] y determinados adenocarcinomas de pulmón, en especial los semisólidos o no sólidos), la captación de 18F-FDG no es la adecuada para obtener imágenes de buena calidad6. Por último, existen muchas dianas relevantes en oncología que no pueden ser evaluadas por la 18F-FDG, como son el transporte de aminoácidos, la síntesis de ADN, la hipoxia, los receptores celulares y la angiogénesis6. Por todo ello, en los últimos años se han desarrollado otros radiofármacos PET, siendo los marcados con 18F los que más han destacado por las ventajas que presenta este radionúclido, sin olvidar los marcados con 68Ga, que están experimentando una gran expansión.
18F-FCH: 18F-colina: fluorometil-dimetil-2-hidroxietil-amonioLa 18F-FCH es un análogo de la colina, compuesto de amonio cuaternario precursor de la fosfatidilcolina, principal fosfolípido de la membrana celular (fig. 1). Las células tumorales presentan un aumento en la biosíntesis de las membranas, por lo que la captación de 18F-FCH proporciona una medida indirecta de la proliferación celular6. Este radiofármaco entra en la célula a través de un transportador, es fosforilado por la colina cinasa y queda retenido en la membrana celular4,6,13,15.
Inicialmente la colina se marcó con 11C, dando lugar a un radiofármaco que, debido a su baja excreción urinaria, presentaba importantes ventajas en las neoplasias urológicas. Sin embargo, su corto T1/2 (20min) suponía un problema logístico si no se disponía de un ciclotrón in situ. Para solucionar este problema de distribución se sintetizó la 18F-FCH14,15.
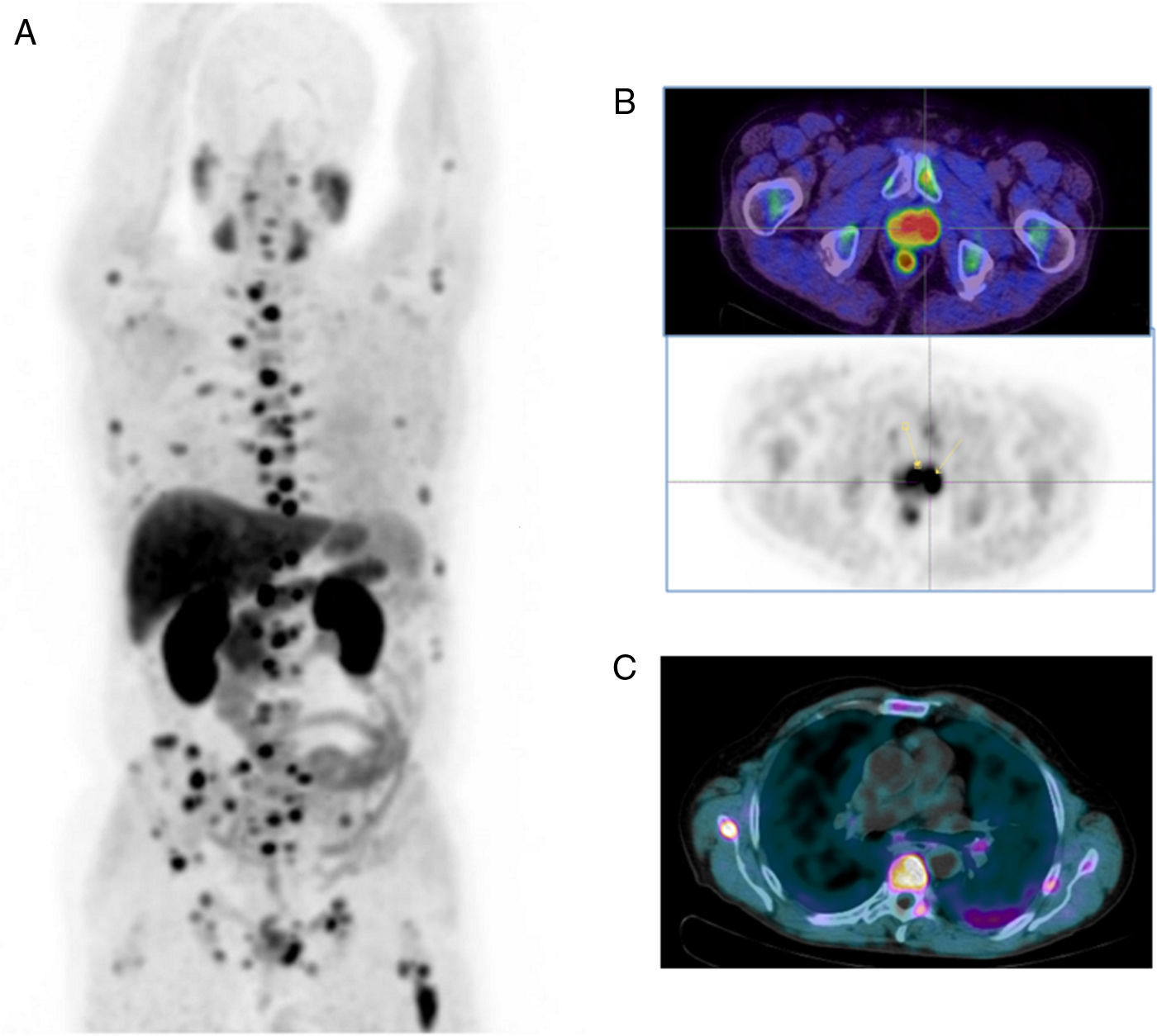
La 18F-FCH está indicada ante la sospecha de tumores cerebrales y de hepatocarcinomas, pero su uso más frecuente es en el diagnóstico precoz de la recidiva del carcinoma prostático tratado con cirugía, radioterapia o ambas, con elevaciones repetidas de las concentraciones séricas de antígeno específico de próstata (PSA) y pruebas de imagen negativas, aunque hay que tener presente que su sensibilidad disminuye cuando el valor del PSA es bajo (≤1-2ng/ml)13,15 (fig. 3). También es importante conocer que los procesos inflamatorios prostáticos y extraprostáticos pueden dar lugar a falsos positivos13.
 (A). Imágenes de fusión PET-TC y PET (B). Imagen de fusión PET-TC (C). Varón de 67 años de edad remitido para estadificación de carcinoma de próstata Gleason 4+3. Se observan varios depósitos patológicos de actividad en la glándula prostática (B), así como múltiples lesiones hipercaptantes en el esqueleto axial y apendicular compatibles con metástasis óseas (A y C).")
Tomografía por emisión de positrones PET-TC con 18F-FCH. Imagen MIP (proyección de máxima intensidad) (A). Imágenes de fusión PET-TC y PET (B). Imagen de fusión PET-TC (C). Varón de 67 años de edad remitido para estadificación de carcinoma de próstata Gleason 4+3. Se observan varios depósitos patológicos de actividad en la glándula prostática (B), así como múltiples lesiones hipercaptantes en el esqueleto axial y apendicular compatibles con metástasis óseas (A y C).
Dadas las limitaciones de la 18F-FDG y de la 18F-FCH para evaluar el cáncer de próstata, en los últimos años se han desarrollado nuevos radiofármacos dirigidos contra dianas específicas de este tumor. El antígeno de membrana específico de próstata (PSMA) es una de las más importantes puesto que está sobreexpresado en el tejido tumoral, de forma que sus valores se correlacionan directamente con la agresividad del tumor, con la independencia de andrógenos, con la enfermedad metastásica y con la progresión de la enfermedad16,17. Se trata de una glucoproteína transmembrana con actividad enzimática en el dominio extracelular que, a diferencia de otros antígenos como el PSA, no es secretada, sino que permanece unida a la membrana, lo que la hace una diana extracelular ideal para el desarrollo de agentes de imagen y terapéuticos18.
Los vehículos necesarios para sintetizar radiofármacos dirigidos contra el PSMA pueden ser de varios tipos, siendo los anticuerpos y los inhibidores del PSMA de bajo peso molecular los más estudiados16. Entre los primeros destaca el anticuerpo monoclonal humanizado J591, específico del dominio extracelular del PSMA, que ha demostrado excelentes características de unión. Su principal desventaja es que presenta una cinética de captación lenta, por lo que la relación óptima tumor/no tumor tarda bastante tiempo en alcanzarse. Por este motivo, debe marcarse con radionúclidos de T1/2 más largo, como el 64Cu (12h) o el 89Zr (78h)16,18. Por el contrario, los inhibidores del PSMA basados en la urea presentan un aclaramiento más rápido. Entre ellos destaca el 68Ga-Glu-urea-Lys(Ahx)-HBED-CC, que según algunos estudios que lo han comparado con la 18F-FCH es capaz de detectar un mayor número de lesiones, en especial cuando los títulos de PSA son bajos19.
18F-FDOPA: L-6-[18F]fluoro-3,4-dihidroxifenilalaninaEs un análogo fluorado de la L-DOPA, aminoácido precursor de la dopamina, la adrenalina y la noradrenalina (fig. 1). Originalmente se desarrolló para estudiar la integridad de la vía dopaminérgica del estriado en pacientes con enfermedad de Parkinson20. También se ha demostrado su utilidad en oncología, ya que al evaluar la actividad de la enzima L-aminoácido aromático descarboxilasa (LAAD), que se encuentra aumentada en los TNE, es un buen biomarcador de este tipo de neoplasias, sobre todo en el feocromocitoma, el carcinoma medular de tiroides y el paraganglioma15,20.
Entre sus indicaciones aprobadas en ficha técnica se encuentran también el diagnóstico y la localización del insulinoma en caso de hiperinsulinismo en lactantes y niños. Permite diferenciar entre enfermedad focal y difusa, lo cual es determinante en el tratamiento y el seguimiento de estos pacientes.
La preparación del paciente puede incluir la premedicación con carbidopa, un inhibidor de la enzima LAAD periférica. Así se consigue que la concentración plasmática de radiofármaco disponible sea mayor y se produce un aumento de su captación, tanto en el estriado como en el caso de los TNE, especialmente en paragangliomas y tumores carcinoides. Sin embargo, no se recomienda el uso de carbidopa en la evaluación de las lesiones pancreáticas, ya que reduce la captación de todo el páncreas y puede dar lugar a falsos negativos21.
68Ga-análogos de la somatostatinaLa somatostatina (SST) es una hormona peptídica cíclica de 14 aminoácidos con un enlace disulfuro entre los residuos 3 y 14 de cisteína, en la cual los aminoácidos situados en los lugares 7, 8, 9 y 10 (fenilalanina, triptófano, lisina, treonina) forman el núcleo activo de la molécula. Está ampliamente distribuida en el organismo, especialmente en el sistema nervioso central y en el sistema nervioso periférico, en el sistema digestivo y en las glándulas endocrinas. Su actividad biológica está mediada por receptores de membrana (SSTR) de alta afinidad, de los que se han descrito cinco subtipos (SSTR1, SSTR2, SSTR3, SSTR4 y SSTR5) con diferente distribución tisular. La SST tiene una acción esencialmente de naturaleza inhibidora o supresora sobre la fisiología normal de diferentes órganos, lo que le confiere efectos beneficiosos en determinadas patologías como los TNE que presentan sobreexpresión de SSTR en su superficie celular22. Sin embargo, no es posible la utilización de SST en la práctica diaria debido, fundamentalmente, a su vida media muy corta (<3min) y al efecto rebote que puede producirse al interrumpir su administración. Por estas razones se han investigado diversos análogos que no sean degradados con tanta rapidez como la SST para, así, tener una vida media más prolongada que permita su uso con fines tanto diagnósticos como terapéuticos22. Entre estos análogos se encuentran la octreotida (OC), la lanreotida y la vapreotida, siendo la primera y sus derivados los más utilizados para la síntesis de radiofármacos.
La imagen basada en análogos de la SST es una herramienta de primera línea para el manejo de los TNE, ya que presentan una serie de ventajas como son su elevada especificidad y su baja antigenicidad, además de poner de manifiesto las dianas que permiten predecir un tratamiento exitoso del tumor con análogos de la SST marcados con itrio-90 (90Y) o lutecio-177 (177Lu)23.
La OC es un péptido de ocho aminoácidos que mantiene el núcleo activo y el puente disulfuro de la SST, mientras que en sus derivados [Tyr3] octreotida (TOC) y [Tyr3] octreotato (TATE) la fenilalanina de la posición 3ha sido sustituida por tirosina, diferenciándose entre ellos en que el carbono terminal de la treonina contiene el ácido carboxílico en el caso del TATE en lugar del grupo alcohol que presenta el TOC. Existe un tercer análogo, la [NaI3] octreotida (NOC), que también se está utilizando como vehículo para la síntesis de radiofármacos PET. Entre los agentes quelantes necesarios para marcar el péptido, con el fin de minimizar la disociación in vivo del radionúclido, destaca el ácido dodecanotetraacético (DOTA), que forma complejos con diversos iones metálicos de gran estabilidad en condiciones fisiológicas24. Se sabe que todas estas variaciones estructurales influyen en las características farmacocinéticas y en el grado de afinidad de los distintos radiofármacos por los diferentes receptores.
Con estos elementos (péptido, quelante y 68Ga) se han sintetizado varios radiofármacos PET, entre los que destacan 68Ga-DOTA-TOC, 68Ga-DOTA-TATE y 68Ga-DOTA-NOC. Aunque los tres pueden unirse a SSTR2, cada uno de ellos tiene sus particularidades en cuanto a su perfil de afinidad por los otros tipos de receptores. Así, 68Ga-DOTA-NOC también presenta una buena afinidad por SSTR3 y SSTR5, y 68Ga-DOTA-TOC por SSTR5, mientras que 68Ga-DOTA-TATE es prácticamente selectivo de SSTR2.
La principal indicación de la PET con 68Ga-análogo de la SST es el estudio de TNE que expresan SSTR. En concreto, se usa para la localización del tumor primario y su estadificación, en el seguimiento de los pacientes para detectar enfermedad residual, recaída o progresión, para determinar la densidad de receptores y seleccionar pacientes con enfermedad metastásica candidatos a recibir tratamiento con péptidos marcados con 90Y o 177Lu 25.
Debido a que estos radiofármacos no tienen aún gran disponibilidad, la gammagrafía y la tomografía por emisión de fotón único (SPECT) de receptores de la SST con 111In-DTPA-octreotida (111In-pentetreotida) sigue siendo la técnica más utilizada para el estudio de los TNE, ya que ha demostrado ser muy útil en su diagnóstico, estadificación y seguimiento, aunque tiene sus limitaciones debido a la gran captación fisiológica en determinados órganos, como el hígado, además de la dificultad para detectar lesiones muy pequeñas debido a la pobre resolución espacial de la propia técnica26,27. La PET con 68Ga-análogos de la SST es superior no solo a la gammagrafía, sino también a la PET con 18F-FDG o con 18F-FDOPA en cuanto a sensibilidad y especificidad, al proporcionar imágenes con un gran contraste gracias a la rápida acumulación de estos radiofármacos en el tumor (80% en 30min) y a su rápido aclaramiento renal, lo que repercute favorablemente en el paciente porque la dosis absorbida es menor y el tiempo de adquisición es más corto7,8.
18F-FLT: 18F-fluorotimidina, 3-[18F]fluoro-3-desoxi-timidinaLa 18F-FLT es un análogo fluorado de la timidina, nucleósido exclusivo del ADN (fig. 1), que una vez ha entrado en el interior de la célula, a través de un sistema transportador, se incorpora rápidamente al ADN tras sufrir tres fosforilaciones, la primera de las cuales es catalizada por la timidina cinasa-18,28.
El primer radiofármaco descrito como indicador de la síntesis de ADN fue la 11C-timidina, en 1972. Sin embargo, su uso no se extendió debido, principalmente, a su corto T1/2, y a que al comportarse de forma idéntica a la timidina también era catabolizada por la timidina fosforilasa dando lugar a metabolitos marcados con 11C que dificultaban de manera importante el análisis de las imágenes obtenidas. Para evitar estos inconvenientes, se sintetizó la 18F-FLT, que actúa como un falso sustrato de la timidina cinasa-1 de forma que, una vez fosforilada, no puede atravesar la membrana, y además, al ser resistente a la degradación, queda atrapada en el interior de la célula, de modo que su acumulación es proporcional a la actividad de dicha enzima. Sin embargo, su incorporación al ADN, a diferencia de la timidina, es mínima (<1%)28.
Su aplicación en oncología se basa en el mayor requerimiento de nucleósidos que tienen los tumores debido a que existen más células en fase S en comparación con el tejido sano. Por tanto, la 18F-FLT, al proporcionar una medida de la síntesis de ADN, puede considerarse un indicador no invasivo de proliferación celular4,6,8. La captación de 18F-FLT se correlaciona con otros medidores de proliferación, como el Ki-67, especialmente en tumores cerebrales, de mama y de pulmón28,29.
La 18F-FLT ha demostrado tener una elevada especificidad para diagnosticar malignidad, ya que da lugar a menos falsos positivos que la 18F-FDG, de forma que, por ejemplo, es muy útil para diferenciar si un nódulo pulmonar solitario es maligno o no, evitando los falsos positivos de la 18F-FDG en caso de granulomatosis y enfermedad inflamatoria15. Sin embargo, su sensibilidad es mucho menor que la de la 18F-FDG debido, entre otros factores, a su gran actividad en el hígado, por glucuronidación del radiofármaco, y en la médula ósea, como consecuencia de la activa proliferación de esta29. Todo ello, unido a que la 18F-FLT también puede dar falsos positivos en caso de ganglios linfáticos reactivos por proliferación de linfocitos, hace que su uso para estadificación sea limitado. Por tanto, la aplicación clínica más prometedora de este radiofármaco es la evaluación precoz de la respuesta al tratamiento, incluso tras la administración del primer ciclo de quimioterapia, ya que la disminución de la proliferación celular constatada con 18F-FLT, en el caso de tumores respondedores, es anterior al descenso de la actividad metabólica medida con 18F-FDG y a la reducción del tamaño medido por TC o resonancia magnética30.
18F-FNa: 18fluoruro sódicoEs un radiofármaco que, debido a los problemas existentes en todo el mundo de disponibilidad de molibdeno-99 y la consecuente escasez de generadores de tecnecio-99m, puede convertirse en una alternativa a la gammagrafía con 99mTc-difosfonatos para la evaluación del sistema óseo, teniendo en cuenta además las ventajas propias de la PET frente a la gammagrafía ósea convencional9,31.
Su mecanismo de captación es similar al de los difosfonatos, pero a diferencia de ellos, el 18F-FNa apenas se une a las proteínas plasmáticas, por lo que su aclaramiento sanguíneo es bastante más rápido. Atraviesa los capilares hacia el fluido extracelular óseo, donde el ion fluoruro se intercambia por un grupo hidroxilo (–OH) de la hidroxiapatita, presente en la superficie de los cristales del hueso, para dar lugar a fluorapatita, principalmente en los sitios con una alta tasa de remodelación ósea32. Estas características farmacocinéticas y su rápida captación ósea hacen que se alcance una buena relación hueso/fondo en poco tiempo, lo que permite la obtención de imágenes a los 45-60 minutos tras la administración del radiofármaco31–34. Su indicación en oncología es la detección de la enfermedad ósea maligna, incluyendo los tumores óseos primarios y las metástasis óseas derivadas de otros tumores4,31.
11C-MET: L-[metil-11C]metioninaLos aminoácidos se usan como vehículo para la síntesis de radiofármacos PET con indicación en oncología, dado que las células tumorales sobreexpresan transportadores de aminoácidos debido a que existe un aumento de su utilización para sintetizar proteínas y otros polipéptidos necesarios para la proliferación celular que tiene lugar durante el crecimiento tumoral4,6.
La L-metionina es un aminoácido esencial cuyas principales funciones son la síntesis de proteínas y la conversión a S-adenosil-metionina, necesaria en numerosas vías metabólicas. Un aumento de la captación de 11C-MET refleja un incremento de transportadores y un aumento de la permeabilidad vascular y de la síntesis de proteínas en el tejido tumoral15.
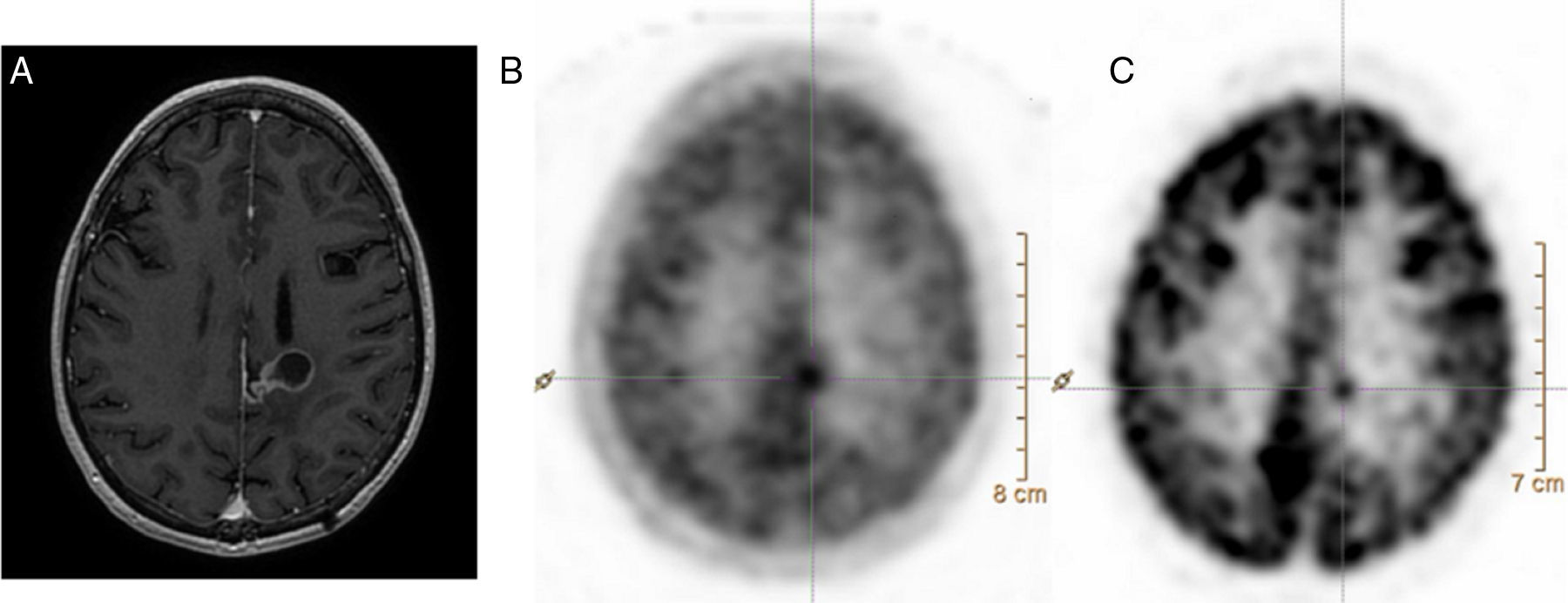
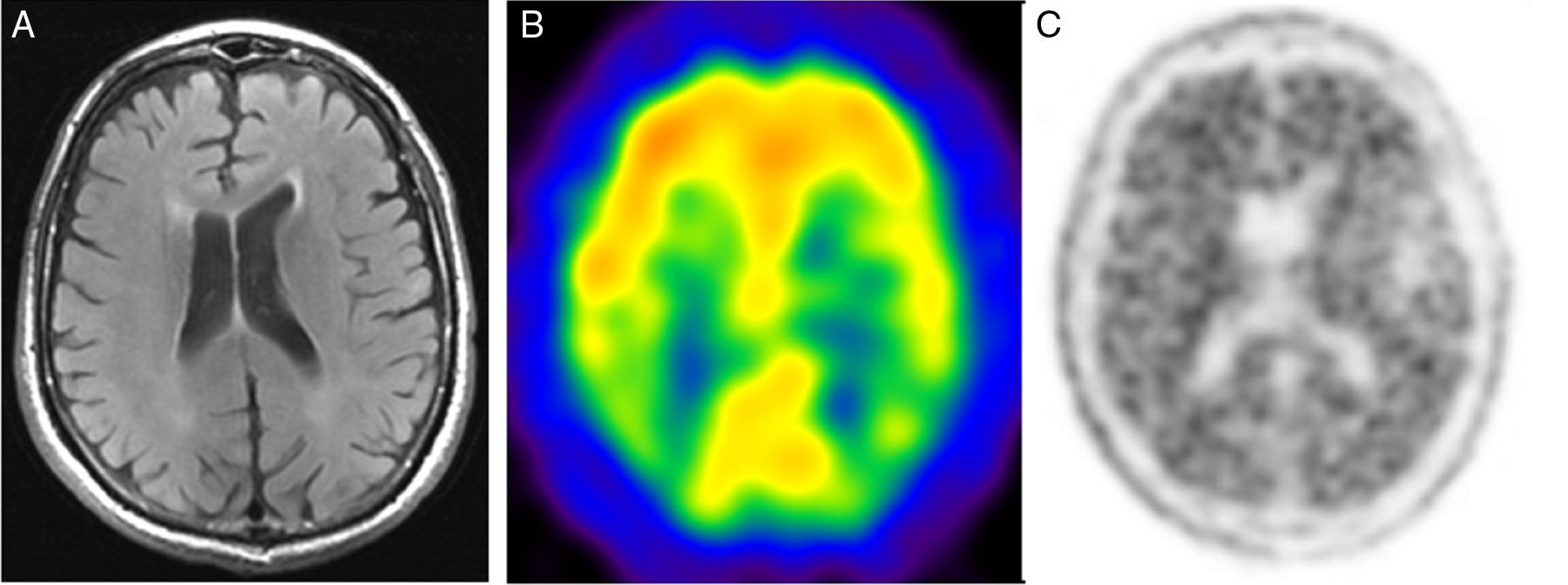
Su principal indicación es el estudio de tumores cerebrales, en los cuales la 18F-FDG es poco sensible, ya que las imágenes obtenidas con 11C-MET presentan una relación tumor/no tumor óptima gracias a la baja captación de aminoácidos en el tejido cerebral normal (fig. 4). Entre sus aplicaciones cabe destacar la discriminación entre recaída y radionecrosis, la identificación de una masa tumoral residual tras la resección, la planificación de la cirugía y la radioterapia, y la posibilidad de servir de guía para realizar una biopsia.
 y cortes axiales de tomografía por emisión de positrones con 11C-MET (B) y con 18F-FDG (C). Mujer de 41 años de edad remitida para realizar diagnóstico diferencial entre recidiva y radionecrosis de glioma cerebral. En la imagen A se aprecia una lesión sólido-quística adyacente al asta occipital del ventrículo lateral izquierdo con realce periférico tras la administración de gadolinio. En los estudios B y C se observa un depósito de actividad en el polo sólido de la lesión ya conocida, compatible con una recidiva tumoral.")
Corte axial de resonancia magnética cerebral en T1 con contraste (A) y cortes axiales de tomografía por emisión de positrones con 11C-MET (B) y con 18F-FDG (C). Mujer de 41 años de edad remitida para realizar diagnóstico diferencial entre recidiva y radionecrosis de glioma cerebral. En la imagen A se aprecia una lesión sólido-quística adyacente al asta occipital del ventrículo lateral izquierdo con realce periférico tras la administración de gadolinio. En los estudios B y C se observa un depósito de actividad en el polo sólido de la lesión ya conocida, compatible con una recidiva tumoral.
Debido a que la 11C-MET sufre metabolismo y da lugar a metabolitos marcados que dificultan el análisis, y a que tiene un T1/2 muy pequeño, se han propuesto otros aminoácidos marcados con 18F, como la 18F-fluoroetiltirosina (18F-FET), cuya captación se correlaciona muy bien con la de 11C-MET porque comparten el mismo sistema de transporte35.
18F-FMISO: 18F-fluoromisonidazol (fig. 1)Es el radiofármaco más estudiado para evaluar de forma no invasiva la hipoxia presente en la mayoría de tumores sólidos. La detección de hipoxia es de gran relevancia clínica, ya que es clave en la progresión y en la resistencia al tratamiento al estar relacionada con la agresividad del tumor, la diseminación metastásica y el aumento de la tasa de recurrencias36.
La imagen PET con 18F-FMISO permite, a diferencia de los métodos invasivos, estudiar todo el tejido tumoral y detectar áreas hipóxicas inaccesibles con dichos procedimientos37.
Su mecanismo de acción se basa en que, una vez atraviesa la membrana celular por difusión pasiva gracias a su lipofilia, es reducido por la enzima nitrorreductasa, de modo que, si la concentración de oxígeno es normal, la molécula es reoxidada y puede volver al espacio extracelular, mientras que, en situación de hipoxia, cuando la PO2 es inferior a 10mmHg, la reducción continúa lentamente hasta unirse de manera covalente a moléculas intracelulares y quedar atrapado dentro de la célula37.
Su principal indicación es la planificación de la radioterapia, debido a que, al identificar las zonas hipóxicas del tumor más resistentes a la radioterapia, pueden sobretratarse respecto a las no hipóxicas38.
Su inconveniente es que presenta una farmacocinética de captación y un aclaramiento lentos, de modo que, para obtener una imagen de calidad, deben pasar entre 2 y 4 horas tras la inyección, lo que dificulta su utilización sistemática38. Por ello se han investigado otros radiofármacos, como el 18F-FAZA (18F-fluoroazomicina arabinósido), el 18F-FETNIM (18F-fluoroeritronitroimidazol) y el 64Cu-ATSM (64Cu-diacetil-bis8N4-metiltiosemicarbazona), que presentan propiedades farmacocinéticas más favorables, aunque la dosis de radiación proporcionada por el 64Cu-ATSM es dos veces la calculada para el 18F-FMISO37.
18F-galacto-RGDEs uno de los primeros radiofármacos desarrollados para el estudio de la angiogénesis inducida por un tumor. En este proceso de neovascularización participan las integrinas, familia de glucoproteínas de membrana formadas por dos subunidades, alfa y beta, que intervienen en las interacciones célula-célula y célula-matriz extracelular. En concreto, el receptor αvβ3 es una de las integrinas, junto con la αvβ5, más importantes en la formación de nuevos vasos y en la producción de metástasis. De ahí su relevancia como objetivo de muchas estrategias terapéuticas y del desarrollo de radiofármacos que permitan obtener imágenes de este proceso y, a su vez, evaluar la respuesta al tratamiento antiangiogénico39.
La estructura química del 18F-galacto-RGD se basa en dos elementos principales (fig. 1). El primero de ellos es la secuencia RGD de tres aminoácidos (arginina-glicina-ácido aspártico), que es reconocida por la integrina αvβ3, lo que le proporciona una gran selectividad. El segundo se refiere a un derivado de la galactosa que mejora las características farmacocinéticas, de modo que las imágenes obtenidas presentan una muy buena relación tumor/fondo40.
Otro radiofármaco desarrollado con el mismo objetivo diagnóstico es la 18F-fluciclatida, que también incluye la secuencia RGD, pero con algunas variaciones que permiten reducir el tiempo de síntesis, lo que haría más factible su producción, además de aumentar su estabilidad frente a la degradación in vivo. A diferencia del anterior, la 18F-fluciclatida tiene más afinidad por la integrina αvβ5 que por la αvβ339
Radiofármacos PET en cardiologíaLa imagen molecular ha contribuido a conocer y entender mejor la patogénesis de algunas enfermedades cardiovasculares, además de detectar, de forma precoz, la progresión de la enfermedad. También cabe destacar su participación en la monitorización de los tratamientos ya existentes, así como en la investigación de nuevas dianas terapéuticas41,42.
Una de las principales aplicaciones de la imagen PET en cardiología es la evaluación de la perfusión miocárdica, aunque debido a la limitada disponibilidad de los radiofármacos PET existentes, bien por la necesidad de tener el ciclotrón in situ (15O-agua y 13N-amoniaco) o por el alto coste del generador, como en el caso del 82Rb, la SPECT sigue siendo la técnica más utilizada y probablemente seguirá siéndolo en la mayoría de los centros. Sin embargo, se espera que, a corto-medio plazo, el uso de la PET crezca de manera significativa gracias, fundamentalmente, al desarrollo de nuevos radiofármacos marcados con 18F, con lo que se ganaría en sensibilidad y exactitud diagnóstica debido a la mayor resolución espacial y temporal de la PET respecto a la SPECT43.
El radiofármaco ideal para realizar estudios de perfusión miocárdica debe presentar una serie de características, entre las que destacan una elevada y rápida captación miocárdica que se correlacione linealmente con el flujo sanguíneo, una baja captación extracardiaca y unas características físicas (energía y T1/2) apropiadas para obtener imágenes de buena calidad y compatibles con la realización de la exploración en reposo y en esfuerzo44. Entre los radiofármacos antes mencionados, el que presenta mayor tasa de extracción miocárdica (100%) es el H215O, es decir, la diferencia en la captación del radiofármaco es directamente proporcional a la diferencia de flujo. Sin embargo, su uso se limita fundamentalmente al cálculo del flujo coronario en investigación, debido a la baja relación miocardio/fondo43,45. El 13NH3 tiene una buena tasa de extracción (85%)45, pero su valor como marcador de perfusión miocárdica está limitado debido a que su captación no depende solo del flujo sanguíneo, sino también de otros factores como son la viabilidad celular, las condiciones metabólicas y la integridad de la membrana celular46. Por último, la tasa de extracción del 82Rb es la más baja (65%), lo cual, unido a su alta energía y su T1/2 ultracorto, hace que no sea muy útil para la cuantificación del flujo sanguíneo miocárdico45,47.
En los últimos años se está investigando un nuevo radiofármaco marcado con 18F. Se trata del 18F-flurpiridaz, análogo del piridaben, que actúa como inhibidor del complejo mitocondrial-1 de la cadena de transporte electrónico, al que se une con gran afinidad. Según los primeros resultados de los estudios de fase 3, expuestos en la Conferencia Internacional de Cardiología Nuclear y Cardio-CT celebrada en Madrid en mayo de 2015, se confirma la evidencia sobre el 18F-flurpiridaz como nuevo radiofármaco para diagnóstico y caracterización de la cardiopatía isquémica. Se trata de un estudio multicéntrico que incluye 795 pacientes con enfermedad coronaria sospechada o confirmada, en el cual uno de los objetivos era comparar la precisión diagnóstica de la PET con 18F-flurpiridaz frente a la SPECT mediante curvas ROC, y que ha demostrado la superioridad de la primera en especial en mujeres, pacientes obesos y pacientes con enfermedad multivaso.
Otra aplicación de la PET en cardiología es la evaluación de la viabilidad miocárdica con 18F-FDG, ya que al ser un marcador del metabolismo permite saber si el tejido miocárdico previamente identificado como hipoperfundido con técnicas de imagen de flujo sanguíneo apropiadas es viable o no, es decir, si capta o no 18F-FDG. Estos estudios son importantes para diferenciar los pacientes que pueden beneficiarse de una revascularización de los que no son candidatos a este tipo de cirugía de alto riesgo47. Para maximizar la captación de 18F-FDG hay que conseguir un estado de hiperinsulinismo (clamp hiperinsulinémico euglucémico, sobrecarga oral de glucosa), de forma que, al aumentar la concentración plasmática de insulina, se favorezca el consumo de 18F-FDG por parte del músculo cardiaco. En caso de haber un importante defecto de perfusión (isquemia grave o necrosis), si el tejido es viable el consumo de 18F-FDG está preservado o aumentado (defecto discordante), mientras que cuando el miocardio hipoperfundido no capta 18F-FDG se habla de miocardio no viable (defecto concordante)47,48.
Por último, la PET también es útil en el estudio de procesos infecciosos e inflamatorios. El diagnóstico de infección de un dispositivo cardiaco o de una prótesis valvular, y especialmente la definición de su localización y extensión, es clave para el tratamiento del paciente, ya que un diagnóstico no concluyente puede llevar a un retraso en el tratamiento antibiótico, la extracción del dispositivo o una intervención quirúrgica con las correspondientes consecuencias que estas situaciones pueden tener para el paciente49. En el caso de procesos inflamatorios, como la sarcoidosis cardiaca, su detección de forma sensible y precisa es fundamental para instaurar lo antes posible una pauta terapéutica con el objetivo de disminuir la morbilidad y la mortalidad asociadas a esta patología50. En ambos casos, la 18F-FDG puede ser de gran ayuda al ser capaz de detectar precozmente la presencia de células inflamatorias (macrófagos, neutrófilos y linfocitos) en la zona de infección o inflamación49. En el caso concreto de la sarcoidosis, permite identificar afectación cardiaca y no cardiaca, además de servir de guía para realizar la biopsia y monitorizar el tratamiento51.
A diferencia de lo que ocurre en la imagen de viabilidad miocárdica, en la cual los hallazgos patológicos son defectos de captación, en los casos de infección o inflamación ocurre lo contrario, ya que es la captación de 18F-FDG la que detecta la presencia de un foco infeccioso o inflamatorio, por lo que es preciso frenar la captación fisiológica del miocardio para su correcta identificación (fig. 5). Para ello, aunque actualmente no existe un protocolo consensuado, algunas estrategias utilizadas incluyen ayuno prolongado (>12h), dietas pobres en hidratos de carbono y ricas en grasas, o la administración de heparina no fraccionada 15 minutos antes de la 18F-FDG con el fin de aumentar las concentraciones plasmáticas de ácidos grasos libres por activación de las lipoproteínas lipasas50,51.
 y corte axial reorientado de la fusión PET-TC (B). Mujer de 69 años de edad con sospecha de endocarditis de válvula mitral. Se observa una intensa actividad metabólica en la válvula protésica aórtica. Captación fisiológica del radiofármaco en cuerpo vertebral.")
Tomografía por emisión de positrones PET-TC con 18F-FDG. Corte axial reorientado de PET (A) y corte axial reorientado de la fusión PET-TC (B). Mujer de 69 años de edad con sospecha de endocarditis de válvula mitral. Se observa una intensa actividad metabólica en la válvula protésica aórtica. Captación fisiológica del radiofármaco en cuerpo vertebral.
La neuroimagen por PET está en expansión debido, entre otros motivos, al desarrollo de radiofármacos que permiten evaluar de forma no invasiva diferentes dianas cerebrales potencialmente disponibles, como son receptores, transportadores, enzimas extracelulares y macromoléculas intracelulares52. El radiofármaco ideal para neuroimagen debe ser de bajo peso molecular, neutro y lipófilo para atravesar la barrera hematoencefálica por difusión pasiva, presentar una alta captación inicial y un rápido aclaramiento, y además tener gran afinidad y selectividad por su diana53,54.
Entre los radiofármacos PET disponibles para la obtención de imágenes cerebrales se encuentran la 18F-FDOPA, la 18F-FDG y los utilizados en la realización de la PET amiloide.
La 18F-FDOPA, como ya se apuntó anteriormente, está indicada en la detección de la pérdida de terminaciones nerviosas dopaminérgicas funcionales del cuerpo estriado en pacientes con síndromes parkinsonianos clínicamente inciertos.
El uso de 18F-FDG en neuroimagen se basa en que es un marcador del metabolismo cerebral. Su principal objetivo diagnóstico es el hipometabolismo glucídico interictal, y su indicación fundamental es la localización de focos epileptógenos en la valoración prequirúrgica de la epilepsia temporal parcial. El hipometabolismo evidenciado por la 18F-FDG también es indicativo de degeneración o daño neuronal subyacente al inicio de la fase de deterioro cognitivo leve y de demencia en la enfermedad de Alzheimer y otras enfermedades neurodegenerativas, mientras que la imagen PET amiloide permite la detección precoz de las fases presintomáticas de la enfermedad de Alzheimer, además de constituir un criterio para excluir otras causas diferentes de esta en el contexto clínico de un deterioro cognitivo leve o una demencia53,55,56.
La PET amiloide se ha convertido, en los últimos años, en un instrumento muy importante para la evaluación de procesos neurodegenerativos que cursan con demencia. Se sabe que un elemento común de muchos de estos procesos es la acumulación patológica de algunas proteínas. En concreto, la enfermedad de Alzheimer se caracteriza por la presencia extracelular de placas neuríticas de β-amiloide y la formación intracelular de ovillos neurofibrilares como consecuencia de la fosforilación de la proteína tau, dando lugar a una degeneración y una pérdida progresivas de neuronas que, finalmente, conducen a atrofia del tejido cerebral54,57,58.
El primer radiofármaco PET amiloide sintetizado fue el 11C-PIB (N-metil-[11C]2-[4-metilaminofenil]-6-hidroxibenzotiazol), también denominado Pittsburgh-Compound-B. Es un análogo de la tioflavina T y es específico para las placas de β-amiloide, con alta afinidad para los compuestos insolubles y baja afinidad para los solubles, siendo la referencia de la imagen cerebral amiloide53. Debido a los inconvenientes derivados del marcaje con 11C ya comentados, se han desarrollado otros radiofármacos marcados con 18F, como son el 18F-flutemetamol, el 18F-florbetapir y el 18F-florbetaben.
El 18F-flutemetamol es también un análogo de la tioflavina que tan solo difiere del 11C-PIB en la presencia del átomo de 18F en posición 3′. Aun cuando sus parámetros cinéticos son favorables, su cinética es más lenta que la de los otros dos radiofármacos fluorados, pero en contrapartida es el que presenta mayor afinidad por la placa amiloide53,58.
El 18F-florbetapir es el que presenta una captación cerebral más rápida, pero menor afinidad por la placa amiloide, mientras que la cinética del 18F-florbetaben es muy similar a la del primero y su afinidad es intermedia entre los dos anteriores59. En cuanto a la especificidad, el 18F-florbetapir es el que tiene un mejor valor (100%), mientras que en sensibilidad es el 18F-florbetaben el que alcanza el máximo valor.
La interpretación de los estudios PET amiloide, a diferencia de los realizados con18F-FDG, se hace mediante una valoración visual estructurada basada en instrucciones detalladas, claras y específicas, que requieren un entrenamiento previo. La interpretación es binaria, es decir, los estudios se clasifican como positivos (densidad alta o moderada de placas corticales de β-amiloide) o negativos (ausencia o densidad escasa de placas corticales de β-amiloide)60 (fig. 6). El estudio PET amiloide debe usarse en combinación con la evaluación clínica, ya que un estudio positivo no equivale a un diagnóstico definitivo de enfermedad de Alzheimer. Sin embargo, la ausencia de placas de β-amiloide sí puede descartar la posibilidad de padecer enfermedad de Alzheimer, lo cual es de notable utilidad en el diagnóstico diferencial.
, corte axial de tomografía por emisión de fotón único con 99mTc-HMPAO (B) y corte axial de tomografía por emisión de positrones con 18F-florbetapir (C). Varón de 74 años de edad con deterioro cognitivo y sospecha de enfermedad de Alzheimer. En la imagen A se observa una pérdida generalizada de masa encefálica sin predominio lobar y signos de leucopatía periventricular laminar gruesa. En el estudio B se identifica hipoperfusión en la región parietooccipital paramedial bilateral, más intensa en el lado izquierdo. En C se aprecia una pérdida de contraste entre la sustancia gris-blanca, compatible con enfermedad de Alzheimer.")
Corte axial de resonancia magnética en T2 y secuencia FLAIR (A), corte axial de tomografía por emisión de fotón único con 99mTc-HMPAO (B) y corte axial de tomografía por emisión de positrones con 18F-florbetapir (C). Varón de 74 años de edad con deterioro cognitivo y sospecha de enfermedad de Alzheimer. En la imagen A se observa una pérdida generalizada de masa encefálica sin predominio lobar y signos de leucopatía periventricular laminar gruesa. En el estudio B se identifica hipoperfusión en la región parietooccipital paramedial bilateral, más intensa en el lado izquierdo. En C se aprecia una pérdida de contraste entre la sustancia gris-blanca, compatible con enfermedad de Alzheimer.
Una de las perspectivas futuras en el campo de la neuroimagen es el desarrollo de nuevos radiofármacos PET cuya diana sea la proteína tau, ya que su acumulación patológica (taupatías) se relaciona con varias enfermedades neurodegenerativas. La imagen tau selectiva permitirá una mejor comprensión de la agregación de tau y su acumulación en el cerebro humano, y proporcionará información sobre las causas, el diagnóstico y el tratamiento de las principales taupatías, tales como la enfermedad de Alzheimer, la encefalopatía traumática crónica, la parálisis supranuclear progresiva, el síndrome corticobasal y algunas variantes de la degeneración frontotemporal lobar61.
ConclusiónHoy por hoy, la oncología sigue siendo el campo de aplicación más importante de la PET y la 18F-FDG es el radiofármaco por excelencia, que influye de manera significativa en la atención del paciente oncológico en cuanto a estadificación, reestadificación y evaluación de la respuesta al tratamiento. Sin embargo, debido a las limitaciones descritas, la 18F-FDG no es útil en todos los tumores, y de ahí la importancia del desarrollo y la comercialización de nuevos radiofármacos PET dirigidos hacia dianas moleculares específicas que amplíen la posibilidad de estudiar otros procesos implicados en el cáncer y otras enfermedades. La estructura de la mayoría de estos nuevos radiofármacos PET se basa, fundamentalmente, en el marcaje con 18F y con 68Ga. El primero de ellos tiene unas características fisicoquímicas muy ventajosas para su producción y distribución, al permitir el suministro de radiofármacos fluorados a centros PET alejados del ciclotrón. Entre ellos destaca la 18F-FLT por su capacidad de evaluar la respuesta al tratamiento de una forma muy precoz, que hace posible optimizar el tratamiento del paciente oncológico. Por otra parte, el 68Ga es un radionúclido con mucho potencial para la síntesis de radiofármacos PET, debido principalmente a su disponibilidad a partir de un generador, siendo de especial interés para el marcaje de péptidos, como es el caso de los 68Ga-análogos de la SST, que permiten obtener imágenes de la densidad de receptores, fundamentales a la hora de plantear el tratamiento de los TNE con péptidos marcados con 90Y o 177Lu. También cabe mencionar los radiofármacos marcados con 68Ga dirigidos hacia el PSMA, ya que se presentan como una alternativa a la 18F-FCH para evaluar el cáncer de próstata, en especial cuando el valor del PSA es bajo.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores han obtenido el consentimiento informado de los pacientes y/o sujetos referidos en el artículo. Este documento obra en poder del autor de correspondencia.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses

