







El rabdomiosarcoma es el sarcoma de partes blandas más frecuente en la edad pediátrica, capaz de aparecer en cualquier parte del organismo. Tiene un comportamiento biológico muy variable y, pese a la ausencia de características clínicas o radiológicas específicas, debe tenerse en cuenta dentro del diagnóstico diferencial de los tumores sólidos en los niños. Esta revisión se centra principalmente en los hallazgos por la imagen y en la distribución anatómica de los subtipos histológicos de rabdomiosarcoma infantil, y de forma secundaria en los hallazgos diferenciales de la anatomía patológica.
Rhabdomyosarcoma is the most common soft-tissue sarcoma in children; it can appear in any part of the body. Its biological behavior varies widely, and despite the absence of specific clinical or radiological characteristics, rhabdomyosarcoma should be taken into account in the differential diagnosis of solid tumors in children. This review focuses primarily on the imaging findings and anatomical distribution of the histological subtypes of childhood rhabdomyosarcoma and secondarily on the differential findings in histological studies.
El rabdomiosarcoma (RMS) es el sarcoma de partes blandas más frecuente en la edad pediátrica y representa un 3-5% de todos los tumores malignos en niños1. Son tumores con un alto potencial de malignidad, originados de las células mesenquimales que posteriormente se diferencian a células de músculo estriado, y pueden aparecer en cualquier parte del organismo, incluso en sitios carentes de esta musculatura2.
De los tres grupos histológicos generales de rabdomiosarcoma, el embrionario (eRMS) representa cerca del 60% de los casos, el alveolar (aRMS) un 30% y el pleomórfico (pRMS), que tiene una frecuencia menor (9-14%), afecta principalmente a los adultos. Existen dos subtipos de eRMS de buen pronóstico, que son el fusiforme, recientemente incluido en la clasificación de la Organización Mundial de la Salud (OMS) como una nueva categoría diagnóstica, y la variante botrioide que ha sido abolida de dicha clasificación3–5.
A pesar de que los subtipos embrionario y alveolar del RMS son predominantemente una patología pediátrica, el alveolar afecta sobre todo a niños mayores o adolescentes y pertenece al grupo pronóstico de alto riesgo según la Clasificación Internacional de Rabdomiosarcoma, mientras que el eRMS es de riesgo intermedio3,4 (tabla 1).
Clasificación histológica del rabdomiosarcoma
| Pronóstico superior | Rabdomiosarcoma embrionario variante fusiforme |
|---|---|
| Pronóstico intermedio | Otras formas de rabdomiosarcoma embrionario |
| Pronóstico pobre | Rabdomiosarcoma alveolar |
| Rabdomiosarcoma indiferenciado | |
| Pronóstico indeterminado | Rabdomiosarcoma con características rabdoides |
La mayoría de los RMS son esporádicos, aunque se cree que un 10-33% tienen un factor de riesgo genético subyacente, con mayor frecuencia síndromes genéticos como el síndrome de Li-Fraumeni, la neurofibromatosis de tipo 1, el síndrome de Beckwith-Wiedemann y el síndrome de Costello4.
Los síntomas son variados e inespecíficos, y aparecen cuando el tumor crece y comprime las estructuras adyacentes, por lo que son dependientes de la localización de la lesión, así como de la presencia o no de diseminación tumoral5. Los pacientes con enfermedad metastásica se presentan con síntomas generales consistentes en cansancio, pérdida de peso y anemia. Una vez se sospecha un tumor de partes blandas, es obligatorio un estudio de imagen con ecografía, tomografía computarizada (TC) o resonancia magnética (RM) para confirmar la sospecha y establecer el estadio, el pronóstico y el protocolo de tratamiento. El diagnóstico definitivo se realiza siempre mediante confirmación histológica; es recomendable que la biopsia se realice en el centro donde se pretende hacer el tratamiento quirúrgico.
Esta revisión se centra principalmente en los hallazgos por imagen y la distribución anatómica de los subtipos histológicos de rabdomiosarcoma infantil, y de forma secundaria en los hallazgos de la anatomía patológica.
HistologíaHistológicamente los RMS pertenecen al grupo de tumores de células azules, pequeñas y redondas de la infancia, grupo en el que también se encuentran el neuroblastoma, el sarcoma de Ewing y el linfoma. Por histología convencional se clasifican mediante el sistema de clasificación internacional de RMS, basado en una relación entre el pronóstico y la histología. Al microscopio óptico, la presencia de miofibrillas y estriaciones cruzadas apunta a la existencia de un tumor esquelético, pero son las técnicas de tinción inmunohistoquímica para marcadores de diferenciación muscular, como la miosina, la desmina, la actina, la miodesmina y la myoD1, las específicas para el rabdomiosarcoma4–6.
Técnicas de imagenLa radiología simple convencional tiene un papel limitado en el diagnóstico de este tipo de tumores y su utilidad está supeditada a la visualización de una tumoración de partes blandas con efecto de masa, o bien a la presencia de signos de infiltración local como erosión ósea. La ecografía tiene una utilidad variable, ya que no solo puede permitir la sospecha diagnóstica ante la presencia de una masa sólida, heterogénea y vascularizada con o sin componente necrótico asociado, sino que dependiendo de la localización de la lesión puede ser útil para realizar una biopsia ecodirigida7. Solo en caso de tumores testiculares la ecografía es la prueba de imagen de elección, aunque también es necesaria una TC de abdomen para evaluar las adenopatías retroperitoneales5.
La TC y la RM, en cambio, son imprescindibles en el diagnóstico y el seguimiento del RMS. La RM se utiliza básicamente para localizar el tumor, estudiar su extensión y valorar signos de invasión local, mientras que la TC está indicada para detectar enfermedad a distancia. Estas técnicas de imagen también se utilizan para evaluar la respuesta tumoral al tratamiento y las recurrencias.
La RM está indicada inicialmente para evaluar el tumor de forma local, incluyendo el área de drenaje en los ganglios linfáticos locorregionales. Idealmente, el estudio debe constar de secuencias basales (FSE-T1 y FSE-T2) para caracterizar la lesión, secuencia de difusión (DWI) y secuencias T1 con y sin contraste con saturación de la grasa para valorar la dimensión tumoral, que debe incluir al menos los dos ejes de mayor longitud, el patrón de realce y los signos de afectación e infiltración de las estructuras adyacentes (afectación de compartimentos musculares contiguos, erosión de la cortical o infiltración de la médula ósea, y compromiso del paquete vasculonervioso, entre otras)8,9. En los pacientes con RMS parameníngeo se recomienda realizar una RM medular para descartar diseminación en el neuroeje6.
Durante el seguimiento de los pacientes, la RM es esencial para valorar la respuesta al tratamiento, ya que aporta información acerca del tamaño tumoral residual y del porcentaje de necrosis tras la quimioterapia (en aquellos en los que está indicada), que se puede valorar mediante la difusión y los mapas de ADC8,9. La RM de cuerpo entero, por su parte, se está utilizando en la actualidad para la detección de metástasis óseas, con una sensibilidad que podría superar a la de la gammagrafía5.
La TC es especialmente útil para detectar signos de afectación e infiltración de las estructuras óseas de difícil valoración por RM, y también debe utilizarse para realizar el estudio de extensión. La detección de metástasis pulmonares mediante TC de tórax sigue los criterios del European paediatric Soft tissue sarcoma Study Group (EpSSG), que incluyen la presencia de un nódulo pulmonar >10mm, o dos o más nódulos bien definidos de 5-10mm, o cinco o más nódulos bien definidos <5mm.
La tomografía por emisión de positrones (PET) con 18-fluorodesoxiglucosa (18-FDG) se usa cada vez más en el diagnóstico y la valoración de la respuesta al tratamiento de los pacientes pediátricos con RMS. No solo detecta el tumor primario y permite visualizar todo el cuerpo en una única sesión, sino que además valora las metástasis regionales, con una rentabilidad diagnóstica equiparable a la de la TC y la RM, y detecta las metástasis a distancia con mayor sensibilidad en comparación con estas técnicas2,5.
En cualquier caso, no hay consenso sobre el uso de PET o RM para la estadificación y el seguimiento de estos pacientes, aunque algunos autores prefieren la RM para minimizar la exposición a la radiación en la población pediátrica.
Hallazgos por la imagenEl RMS se presenta típicamente como una lesión única y unilateral, sólida, expansiva, altamente vascularizada y con un grado variable de componente quístico-necrótico.
En la TC se muestra como una lesión de densidad de partes blandas, de contornos bien definidos y con un patrón de realce poscontraste similar o discretamente hipodenso comparado con el de la musculatura adyacente. En la RM veremos una tumoración de señal intermedia o isointensa en secuencias T1, hiperintensa en secuencias T2 y STIR, con o sin artefactos de vacío de señal, que indica la rica vascularización del tumor. En las secuencias T1 con saturación grasa poscontraste hay un intenso realce del componente sólido, o bien un realce anular en caso de tener un gran componente quístico-necrótico. Estos tumores, debido a su rica celularidad, presentan una alta señal en las secuencias DWI, con caída de señal en los mapas ADC; dicha restricción en la difusión, al igual que el tamaño tumoral, son herramientas fundamentales para valorar la respuesta a la quimioterapia, ya que la disminución de tamaño y la ausencia de restricción en la difusión durante el seguimiento indican una buena respuesta7,10,11.
La PET con 18-FDG es una imagen morfológica y funcional capaz de localizar una masa de partes blandas de similares características a las descritas previamente para la TC, y además permite hacer una valoración cualitativa y cuantitativa de la captación de 18-FDG en el tejido tumoral, que en el caso de los RMS muestra un metabolismo alto. Dicho hipermetabolismo es de utilidad para valorar el grado histológico y guiar la biopsia, y durante el seguimiento puede hacerse una medición comparativa del metabolismo y del tamaño tumoral para evaluar la respuesta al tratamiento, así como para detectar las posibles recurrencias.
Aunque el RMS carece de hallazgos patognomónicos por imagen que permitan hacer un diagnóstico específico, tanto en los casos que presentamos como en la mayoría de los descritos en la literatura es poco frecuente la asociación con calcificaciones, hemorragia o grasa macroscópica.
ClasificaciónTanto el eRMS como el aRMS pueden localizarse indistintamente en cualquier parte del organismo, aunque cada subtipo histológico tiene predilección para localizarse en algunos órganos y sistemas4 (fig. 1). Así:
- •
Subtipo embrionario:
- a)
Cabeza y cuello (parameníngeo, periorbitario, no parameníngeo).
- b)
Tracto genitourinario.
- c)
Retroperitoneo.
- a)
- •
Subtipo alveolar:
- d)
Extremidades.
- e)
Tronco.
- d)

Es el subtipo histológico más frecuente, afecta a pacientes de menor edad y tiene un mejor pronóstico4,12. Sus células tienen un enorme parecido a los distintos estadios de la embriogénesis de las células musculoesqueléticas normales5,6,13.
Aunque no se sabe mucho sobre las causas por las que una célula mesenquimal se desdiferencia en célula muscular esquelética y esta se transforma en célula cancerosa, sí se conocen los cambios genéticos que tienen lugar en los casos de eRMS, en tanto que hay una sobreexpresión del gen IGF-II localizado en el brazo corto del cromosoma 11. Esta expresión de las dos copias del gen causa un efecto de «sobredosis» en la que dicho exceso produce una señal constante de inducción a la proliferación, que permite a las células musculares preneoplásicas (o ya con transformación tumoral) crecer de forma incontrolada14. La evolución natural del RMS varía dependiendo del origen anatómico de la lesión, y por eso cada localización debe ser recordada como una afección propia que difiere en sus características clínicas, patrón de diseminación, tratamiento y pronóstico.
Prácticamente la mitad de los eRMS se originan en la cabeza y el cuello4, y esta localización también es el sitio más frecuente de aparición de un segundo tumor primario (fosa temporal y fosas etmoidales), sobre todo en los supervivientes de un retinoblastoma hereditario que han sido sometidos a radioterapia15.
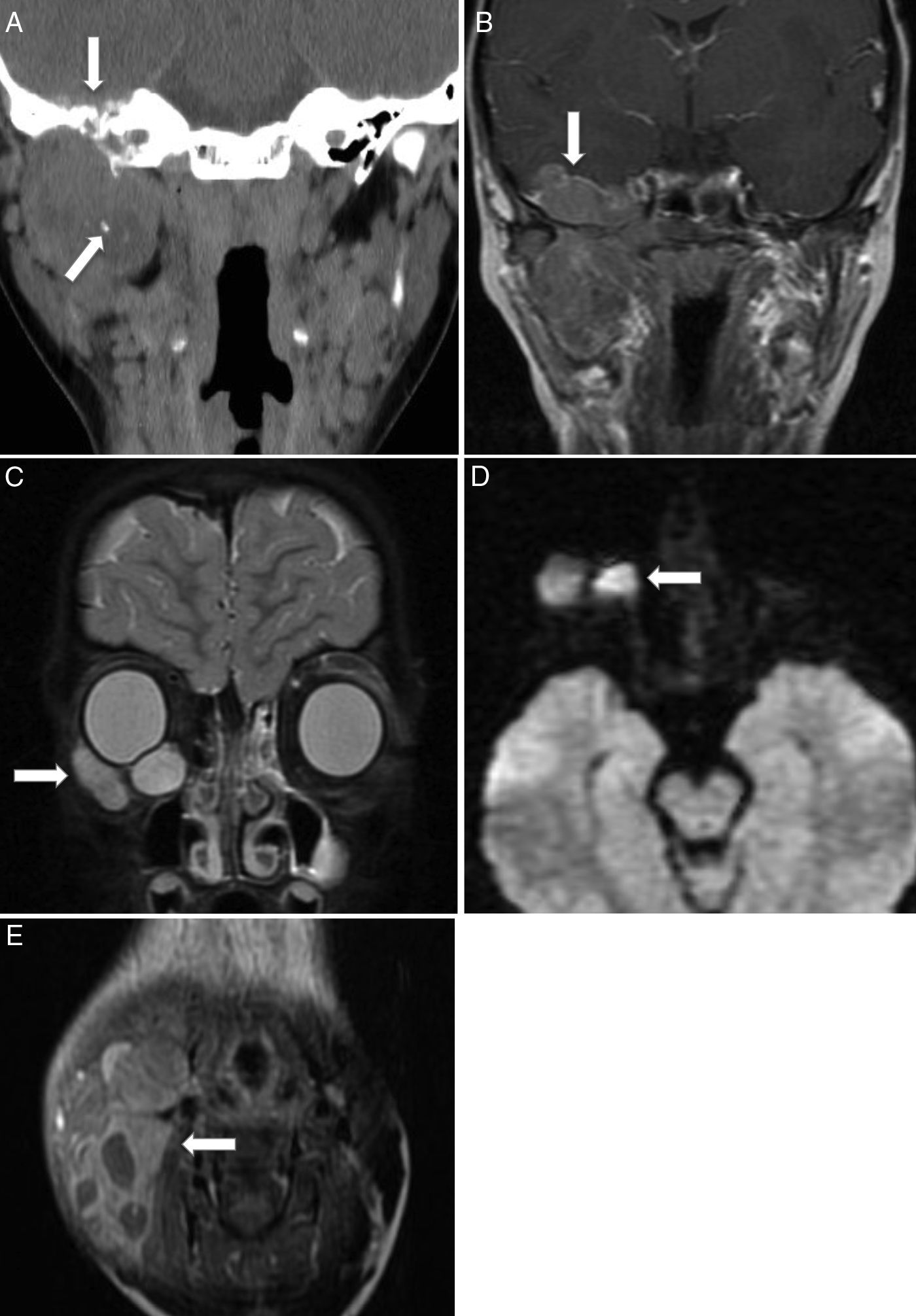
Según su localización, los tumores de cabeza y cuello se dividen en parameníngeos (50%), periorbitarios (25%) y no parameníngeos (20%)2,4. Los tumores localizados en la región parameníngea no orbitaria suelen presentarse con obstrucción nasal o sinusal, rinorrea o parálisis de nervios craneales, lo que sugiere extensión hacia las meninges. Los tumores primarios de órbita suelen presentarse con proptosis u oftalmoplejía, y en general se diagnostican antes de la diseminación tumoral. El resto de los tumores suelen presentarse como masas no dolorosas aumentadas de tamaño, casi siempre localizadas. Las metástasis más frecuentes son en el pulmón y el hueso2,5,6,12. El diagnóstico diferencial en esta localización debe incluir el linfoma, el carcinoma nasofaríngeo, los tumores neuroectodérmicos, la histiocitosis de células de Langerhans, el neuroblastoma olfatorio, el osteosarcoma y las metástasis, entre otros (fig. 2).
 corte coronal de TC facial sin contraste con ventana de partes blandas que muestra una lesión expansiva heterogénea con necrosis central y calcificaciones en la región infratemporal derecha, que erosiona y remodela el hueso de la rama mandibular y el peñasco (flechas); B) corte coronal de RM T1 poscontraste que muestra el realce heterogéneo de la tumoración, con extensión intracraneal al segmento basal del lóbulo temporal (flecha). Rabdomiosarcoma periorbitario: C) corte coronal de RM de órbita STIR que muestra una lesión bien delimitada en el suelo de la órbita derecha (flecha), con comportamiento de señal hiperintenso; D) corte transversal de RM de difusión que muestra restricción de dicha lesión (flecha). Rabdomiosarcoma de cuello: E) corte transversal de RM T1 poscontraste que muestra una tumoración sólida que se expande desde el espacio masticatorio hasta el ángulo mandibular derecho (flecha), con componente quístico-necrótico en su interior y realce heterogéneo de contraste de la porción sólida. Asocia un extenso conglomerado adenopático, que rodea la lesión y es indistinguible de la tumoración primaria.")
Rabdomiosarcoma embrionario de cabeza y cuello. Rabdomiosarcoma parameníngeo: A) corte coronal de TC facial sin contraste con ventana de partes blandas que muestra una lesión expansiva heterogénea con necrosis central y calcificaciones en la región infratemporal derecha, que erosiona y remodela el hueso de la rama mandibular y el peñasco (flechas); B) corte coronal de RM T1 poscontraste que muestra el realce heterogéneo de la tumoración, con extensión intracraneal al segmento basal del lóbulo temporal (flecha). Rabdomiosarcoma periorbitario: C) corte coronal de RM de órbita STIR que muestra una lesión bien delimitada en el suelo de la órbita derecha (flecha), con comportamiento de señal hiperintenso; D) corte transversal de RM de difusión que muestra restricción de dicha lesión (flecha). Rabdomiosarcoma de cuello: E) corte transversal de RM T1 poscontraste que muestra una tumoración sólida que se expande desde el espacio masticatorio hasta el ángulo mandibular derecho (flecha), con componente quístico-necrótico en su interior y realce heterogéneo de contraste de la porción sólida. Asocia un extenso conglomerado adenopático, que rodea la lesión y es indistinguible de la tumoración primaria.
En el tracto genitourinario, la mayoría de los RMS son del subtipo embrionario. Constituyen la segunda localización tras la cabeza y el cuello, y representan un 15-30% de todos los RMS en niños4,5. Se incluyen en este grupo los tumores originados en la vejiga, la próstata, el testículo y la región paratesticular, el pene, el útero, la vagina y la región perineal. Se dividen anatómicamente en los que se originan en la región paratesticular, la vejiga-próstata y ginecológicos, aunque desde el punto de vista pronóstico existe una distinción entre los tumores genitourinarios en vejiga-próstata, de peor pronóstico, y los genitourinarios de otras localizaciones, como los paratesticulares, los uterinos y los vaginales, de mejor pronóstico5.
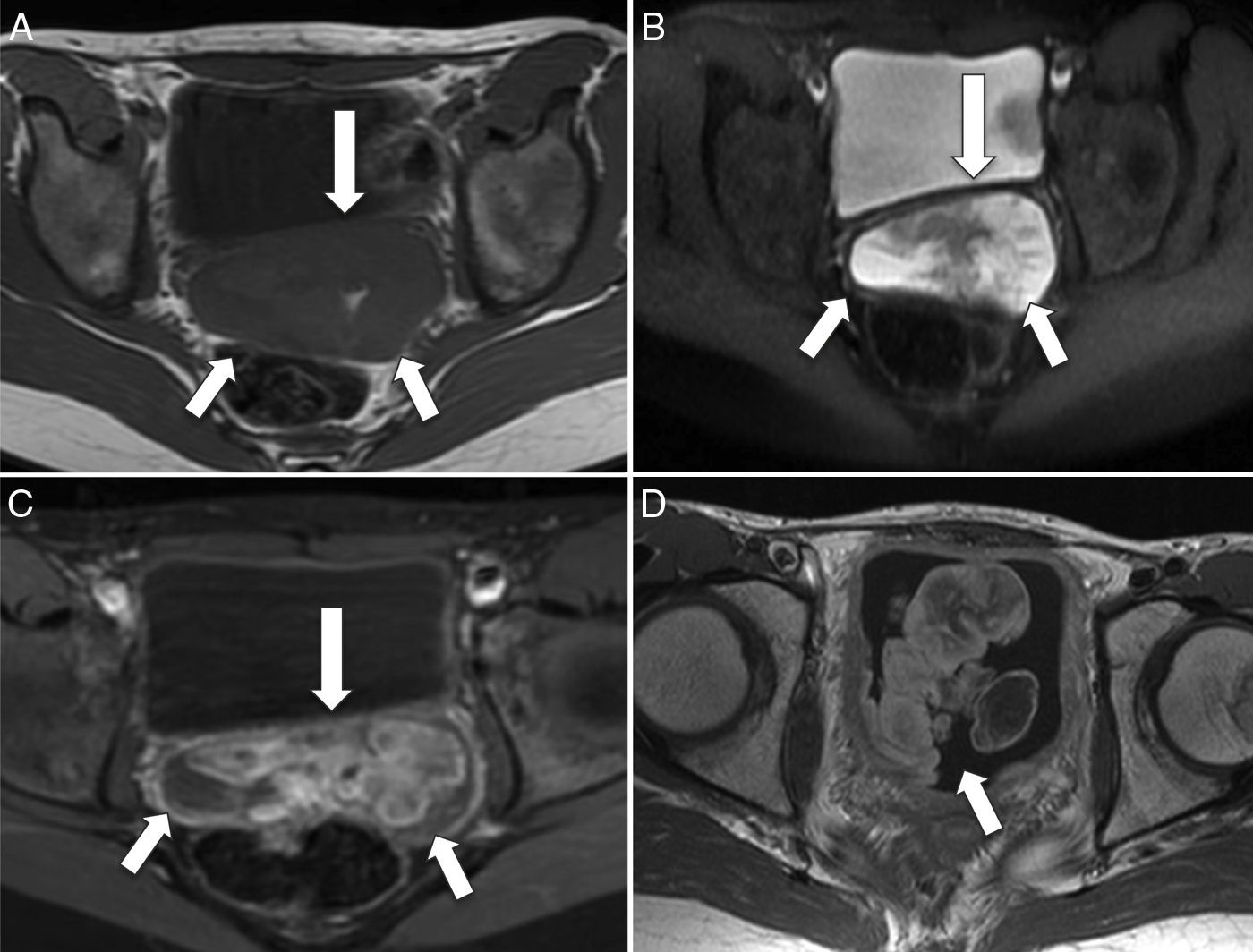
La localización en la vejiga ocurre con frecuencia en niños menores de 4 años y se caracteriza por el crecimiento exofítico intraluminal. El origen prostático, por el contrario, muestra tumoraciones más grandes, y su diseminación al pulmón, la médula ósea y el hueso es más temprana. El RMS vaginal afecta a niñas de muy corta edad y se caracteriza por ser una masa de crecimiento polipoideo intraluminal, similar a la que se origina en la submucosa de órganos huecos como la vejiga, la vagina, la cavidad nasal, la nasofaringe y la vía biliar. Esta morfología es característica de la antiguamente denominada variedad botrioide, abolida de la última clasificación de la OMS. Microscópicamente, dicha variedad se caracteriza por su relativa escasez de células y la abundancia de estroma mucoide, que a menudo resulta similar al mixoma4–6 (fig. 3).
 Corte transversal de RM potenciada en T1 que muestra un aumento de partes blandas hipointenso (flechas), que protruye a través del fondo de saco de Douglas. B) Corte transversal de RM potenciada en T2 que muestra la misma lesión con comportamiento de señal hiperintenso y heterogéneo (flechas). C) Corte transversal T1-FS poscontraste en el que se observa el realce heterogéneo de contraste de la lesión (flechas). D) Corte transversal de RM T1-FS poscontraste en el que se observa una lesión exofítica intravesical (flecha) con realce heterogéneo de contraste, de similares características que la tumoración vaginal.")
Rabdomiosarcoma embrionario de morfología botrioide en la vagina y la vejiga urinaria. A) Corte transversal de RM potenciada en T1 que muestra un aumento de partes blandas hipointenso (flechas), que protruye a través del fondo de saco de Douglas. B) Corte transversal de RM potenciada en T2 que muestra la misma lesión con comportamiento de señal hiperintenso y heterogéneo (flechas). C) Corte transversal T1-FS poscontraste en el que se observa el realce heterogéneo de contraste de la lesión (flechas). D) Corte transversal de RM T1-FS poscontraste en el que se observa una lesión exofítica intravesical (flecha) con realce heterogéneo de contraste, de similares características que la tumoración vaginal.
Otras localizaciones menos típicas son las extremidades, el tronco, la pelvis, el retroperitoneo y el periné4 (figs. 4 y 5).
 Ecografía del área paraespinal derecha que muestra una lesión heterogénea, con centro necrótico-quístico (asterisco), muy vascularizada. B) Corte transversal de TC sin contraste con ventana de partes blandas que muestra una tumoración sólida (flecha) e isodensa con el músculo adyacente, sin signos de afectación costal. C) RM transversal en secuencia T1 en la que la lesión es isointensa a la musculatura (flecha), con posterior realce heterogéneo (flecha) tras la administración de contraste (D).")
Rabdomiosarcoma embrionario paraespinal. A) Ecografía del área paraespinal derecha que muestra una lesión heterogénea, con centro necrótico-quístico (asterisco), muy vascularizada. B) Corte transversal de TC sin contraste con ventana de partes blandas que muestra una tumoración sólida (flecha) e isodensa con el músculo adyacente, sin signos de afectación costal. C) RM transversal en secuencia T1 en la que la lesión es isointensa a la musculatura (flecha), con posterior realce heterogéneo (flecha) tras la administración de contraste (D).
 Ecografía de abdomen que muestra una gran tumoración sólida heterogénea (asterisco), altamente vascularizada. B) Corte transversal de RM T2 que confirma la gran extensión de un tumor heterogéneo (flechas), con múltiples tabiques en su interior. C) Corte transversal de RM T1-FS poscontraste que muestra un realce irregular de la tumoración (flechas) tras la administración de contraste paramagnético.")
Rabdomiosarcoma embrionario abdominal. A) Ecografía de abdomen que muestra una gran tumoración sólida heterogénea (asterisco), altamente vascularizada. B) Corte transversal de RM T2 que confirma la gran extensión de un tumor heterogéneo (flechas), con múltiples tabiques en su interior. C) Corte transversal de RM T1-FS poscontraste que muestra un realce irregular de la tumoración (flechas) tras la administración de contraste paramagnético.
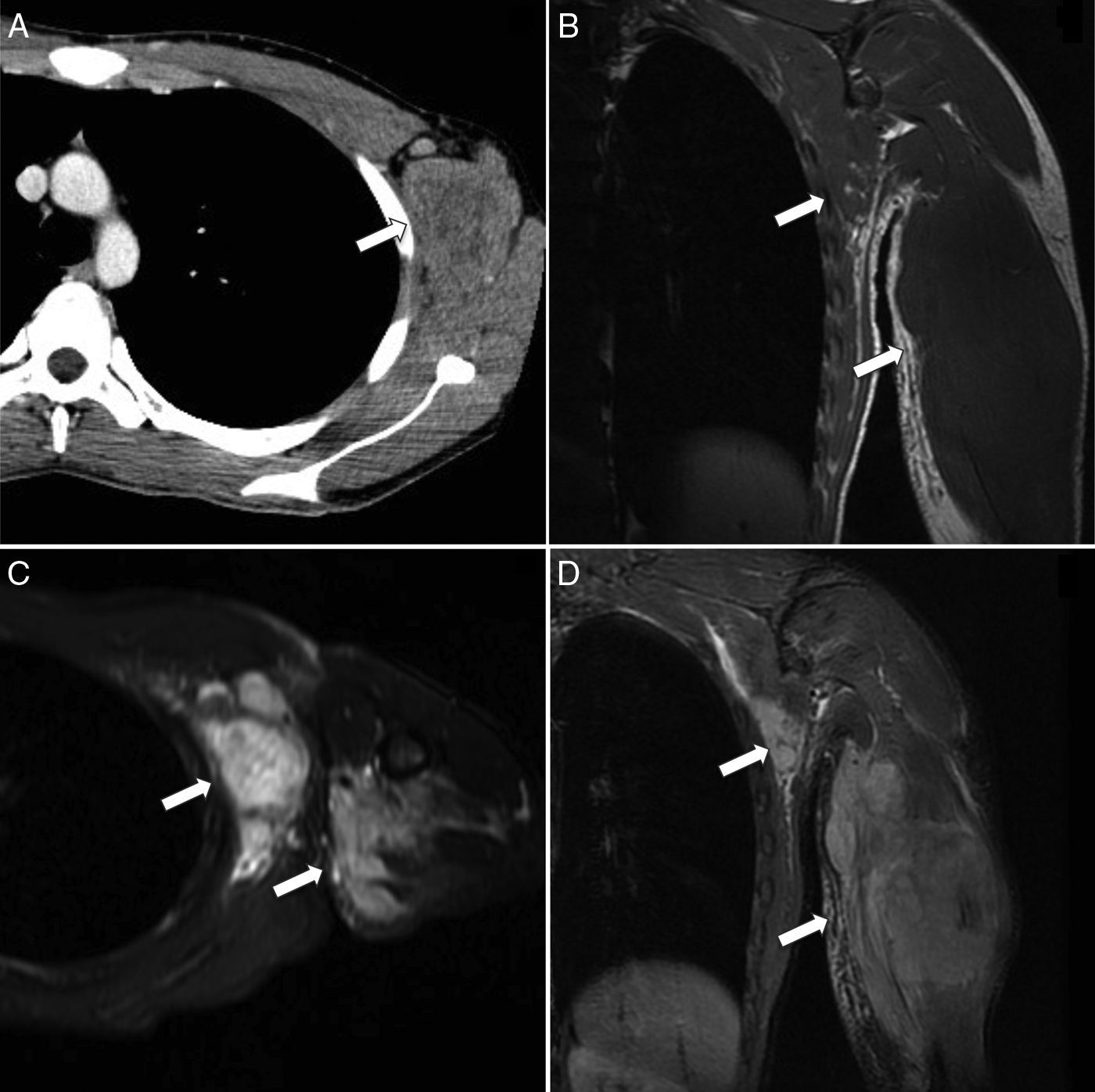
Representa menos del 1% de todos los tumores de tejidos blandos y alrededor del 20-30% de los RMS4–6,10,16. Aunque en el tronco y las extremidades se han descrito todos los subtipos histológicos de RMS, el subtipo alveolar tiene predilección por estas dos localizaciones (fig. 6).
 Corte transversal de TC con contraste que muestra un aumento de volumen heterogéneo de partes blandas en la región axilar y pectoral izquierda (flecha). B) Corte coronal de RM T1 que muestra un aumento de volumen de la musculatura, mal delimitado, debido al comportamiento isointenso de la lesión en relación al músculo normal (flechas). C y D) Corte transversal y coronal de RM T2-FS donde la masa se presenta como una extensa lesión polilobulada e hiperintensa en la pared torácica y el brazo (flechas). Edema del tejido celular subcutáneo asociado.")
Rabdomiosarcoma alveolar de tronco y extremidad superior. A) Corte transversal de TC con contraste que muestra un aumento de volumen heterogéneo de partes blandas en la región axilar y pectoral izquierda (flecha). B) Corte coronal de RM T1 que muestra un aumento de volumen de la musculatura, mal delimitado, debido al comportamiento isointenso de la lesión en relación al músculo normal (flechas). C y D) Corte transversal y coronal de RM T2-FS donde la masa se presenta como una extensa lesión polilobulada e hiperintensa en la pared torácica y el brazo (flechas). Edema del tejido celular subcutáneo asociado.
Debido a su ritmo de crecimiento relativamente indolente, pueden ser tumores de gran extensión en el momento del diagnóstico y tienen un pronóstico menos favorable que el subtipo embrionario.
Se caracteriza por tener una imagen histológica típica y alteraciones moleculares específicas. Así, cerca del 90% de los casos tienen una translocación que involucra uno de los genes PAX (más comúnmente PAX 3, en el cromosoma 2, y con menos frecuencia PAX 7, en el cromosoma 1) y el gen forkhead (FKHR) localizado en el cromosoma 13. Las proteínas de fusión resultantes, PAX3-FKHD y PAX7-FKHD, son específicas de este tipo de tumores y cumplen criterios esenciales para funcionar como factor de transcripción aberrante, facilitando la actividad cancerígena y la proliferación celular anómala4,6,17,18.
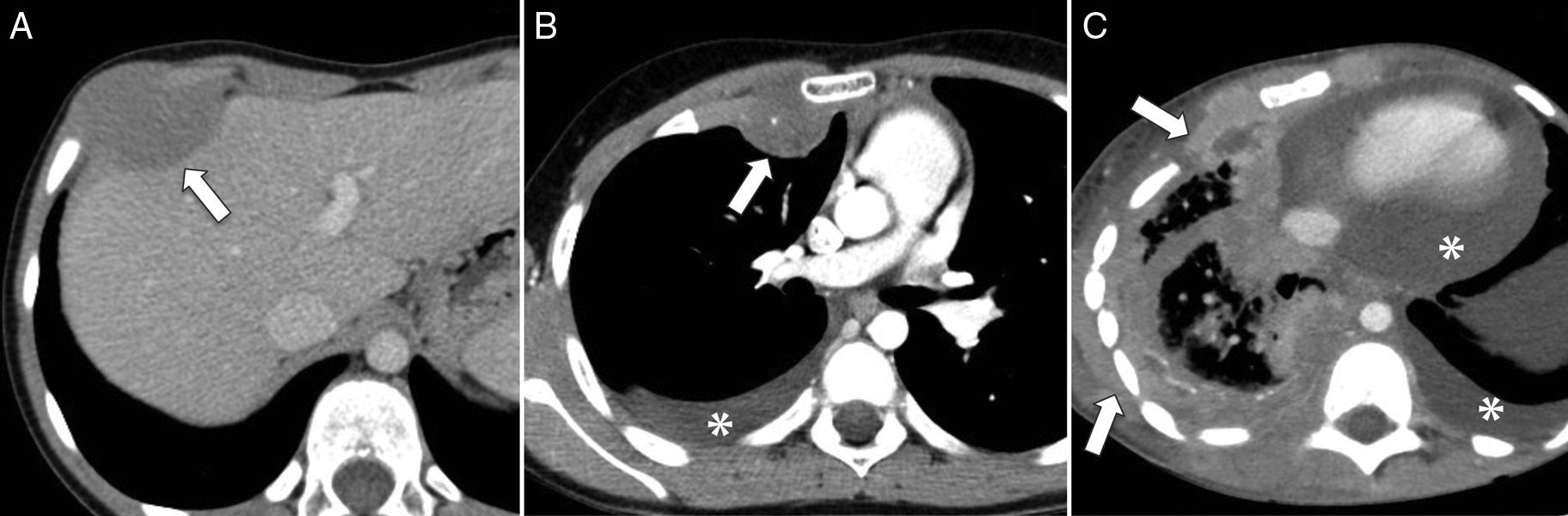
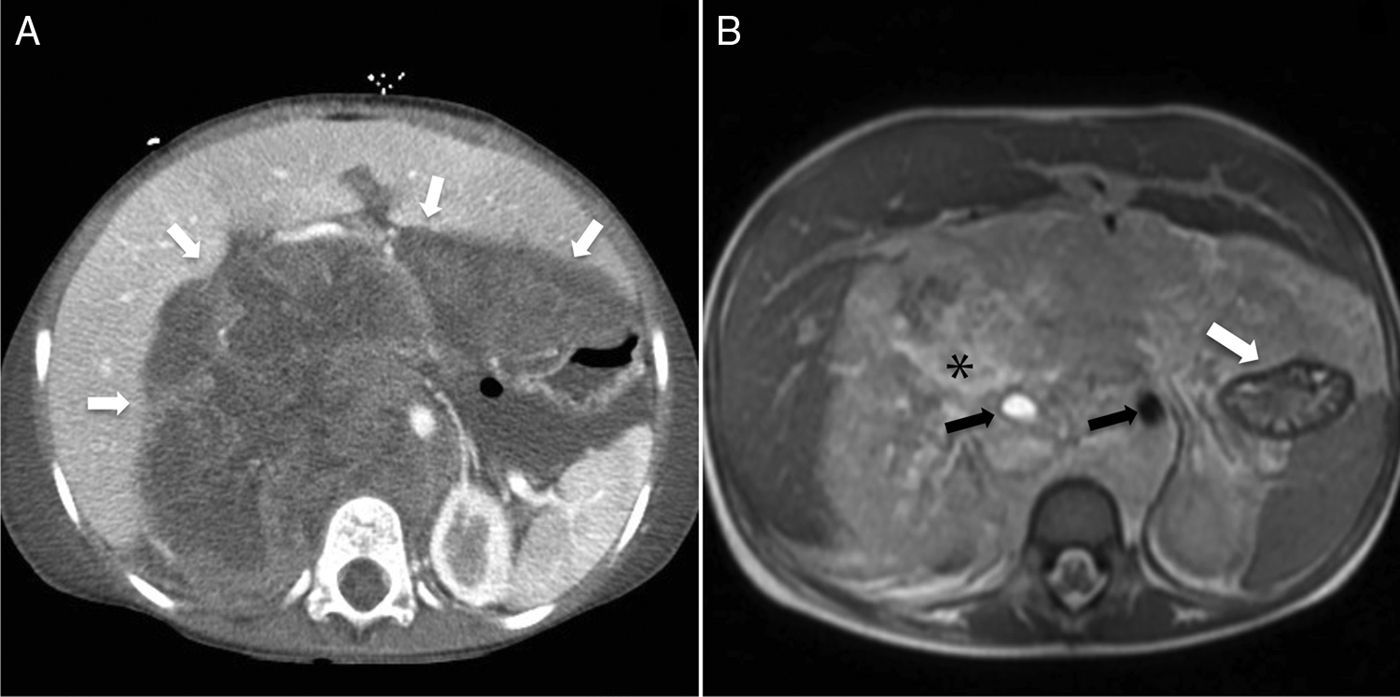
El aRMS tiende a recurrir localmente después de su escisión y hasta un 65% de los pacientes desarrollan enfermedad a distancia11, que en un alto porcentaje es además resistente a la quimioterapia convencional, y de ahí su mal pronóstico (fig. 7). Estas metástasis aparecen primero en el pulmón, el hueso, el sistema nervioso central y el hígado, y en lugares inusuales como la mama10,19 (fig. 8).
 Corte transversal de TC abdominal con contraste intravenoso que muestra una tumoración de partes blandas en la pared torácica anterior derecha (flecha), hipodensa en relación al músculo, que abomba sobre la cara interna de la pared toracoabdominal y remodela el contorno hepático adyacente, sin aparente plano de clivaje. B) Corte transversal de TC torácica con contraste intravenoso durante el seguimiento del mismo paciente que muestra una recaída en la pared torácica (flecha), con derrame pleural homolateral (asterisco). C) Corte transversal de TC torácica con contraste intravenoso meses más tarde, que muestra progresión tumoral con engrosamiento irregular de la pleura e infiltración de la pared torácica (flechas), así como derrame pleural y pericárdico (asteriscos).")
Rabdomiosarcoma alveolar de tronco. A) Corte transversal de TC abdominal con contraste intravenoso que muestra una tumoración de partes blandas en la pared torácica anterior derecha (flecha), hipodensa en relación al músculo, que abomba sobre la cara interna de la pared toracoabdominal y remodela el contorno hepático adyacente, sin aparente plano de clivaje. B) Corte transversal de TC torácica con contraste intravenoso durante el seguimiento del mismo paciente que muestra una recaída en la pared torácica (flecha), con derrame pleural homolateral (asterisco). C) Corte transversal de TC torácica con contraste intravenoso meses más tarde, que muestra progresión tumoral con engrosamiento irregular de la pleura e infiltración de la pared torácica (flechas), así como derrame pleural y pericárdico (asteriscos).
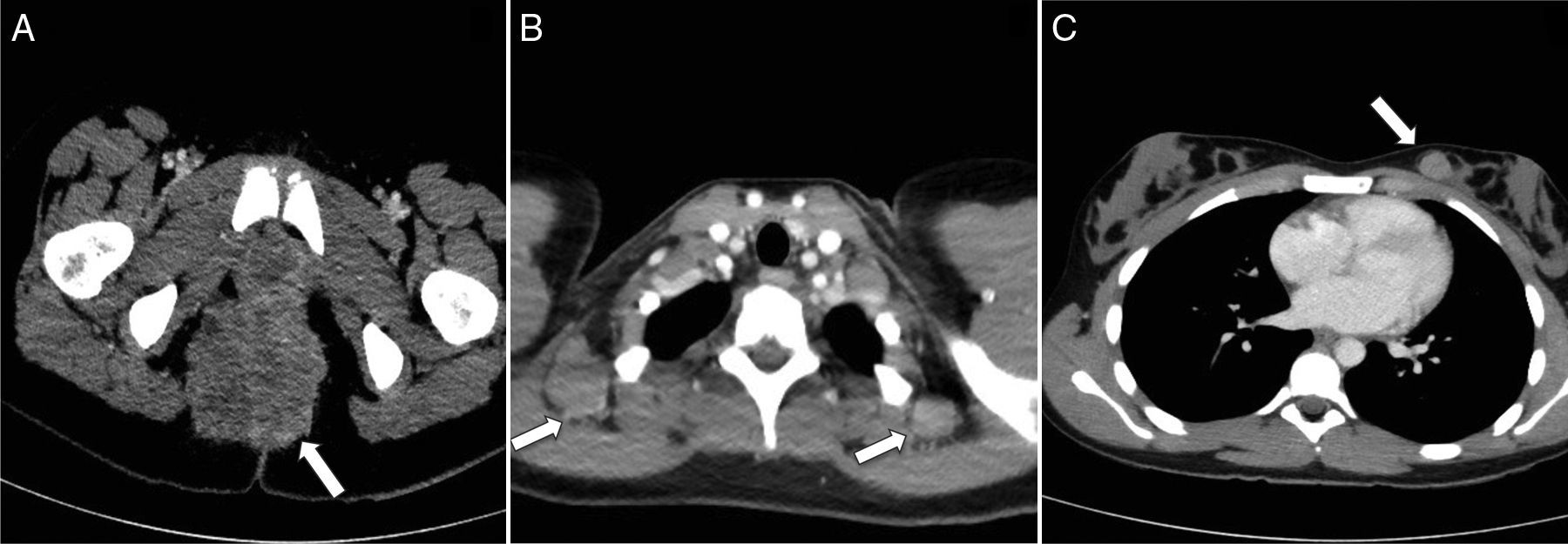
 Corte transversal de TC pélvica con contraste intravenoso que muestra una tumoración heterogénea de contornos lobulados en la región perineal y glútea (flecha). B) Corte transversal de TC torácica con contraste en transición cérvico-torácica con evidencia de diseminación tumoral a los ganglios cervicales posteroinferiores en el nivel VB (flechas). C) En el mismo paciente, la TC torácica muestra metástasis en la mama izquierda (flecha).")
Rabdomiosarcoma alveolar perineal. A) Corte transversal de TC pélvica con contraste intravenoso que muestra una tumoración heterogénea de contornos lobulados en la región perineal y glútea (flecha). B) Corte transversal de TC torácica con contraste en transición cérvico-torácica con evidencia de diseminación tumoral a los ganglios cervicales posteroinferiores en el nivel VB (flechas). C) En el mismo paciente, la TC torácica muestra metástasis en la mama izquierda (flecha).
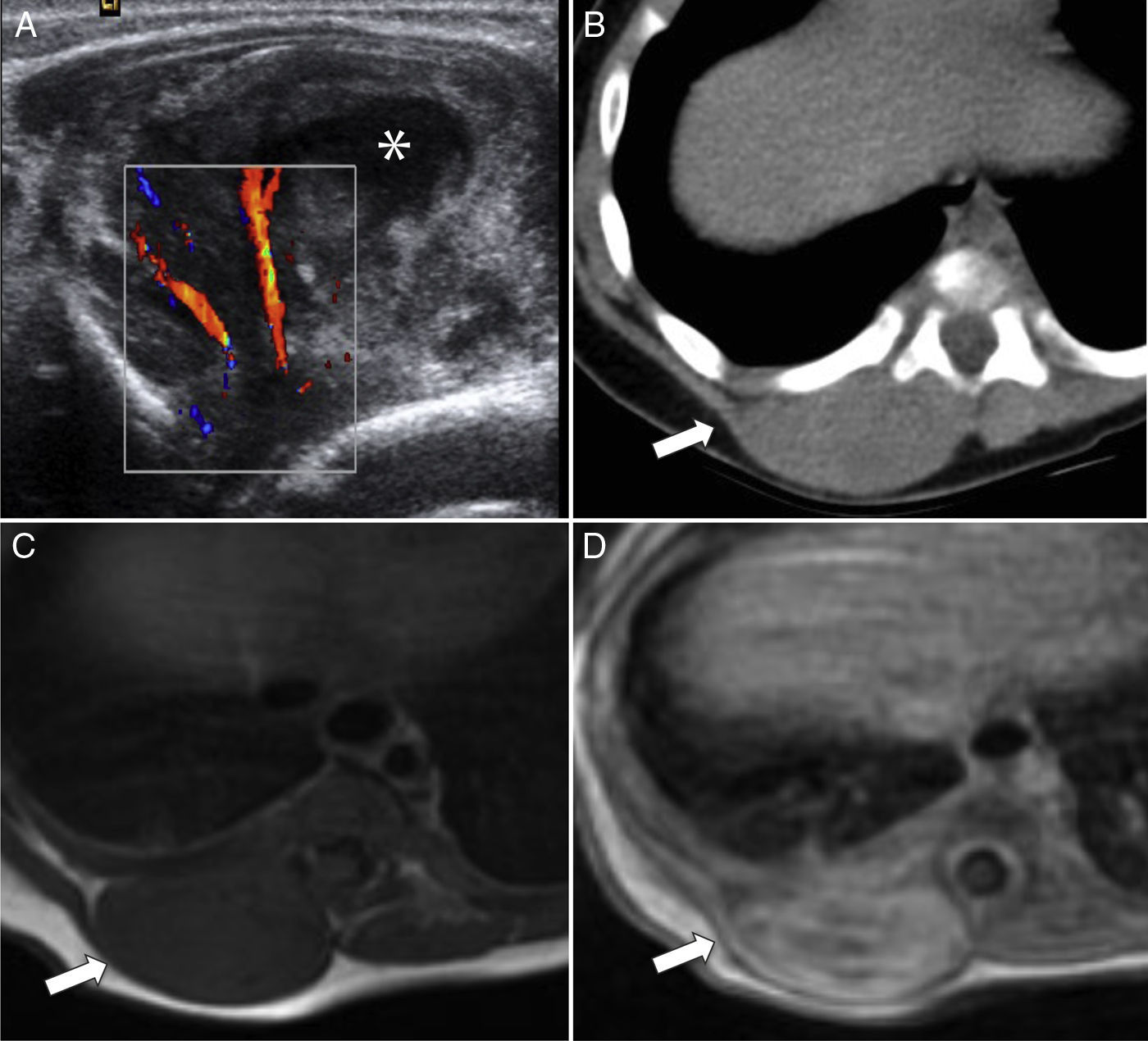
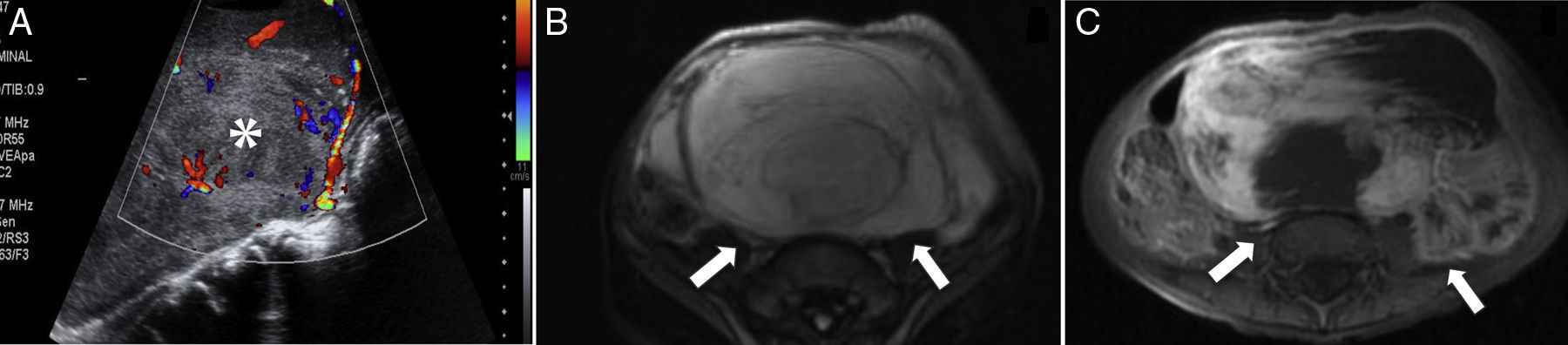
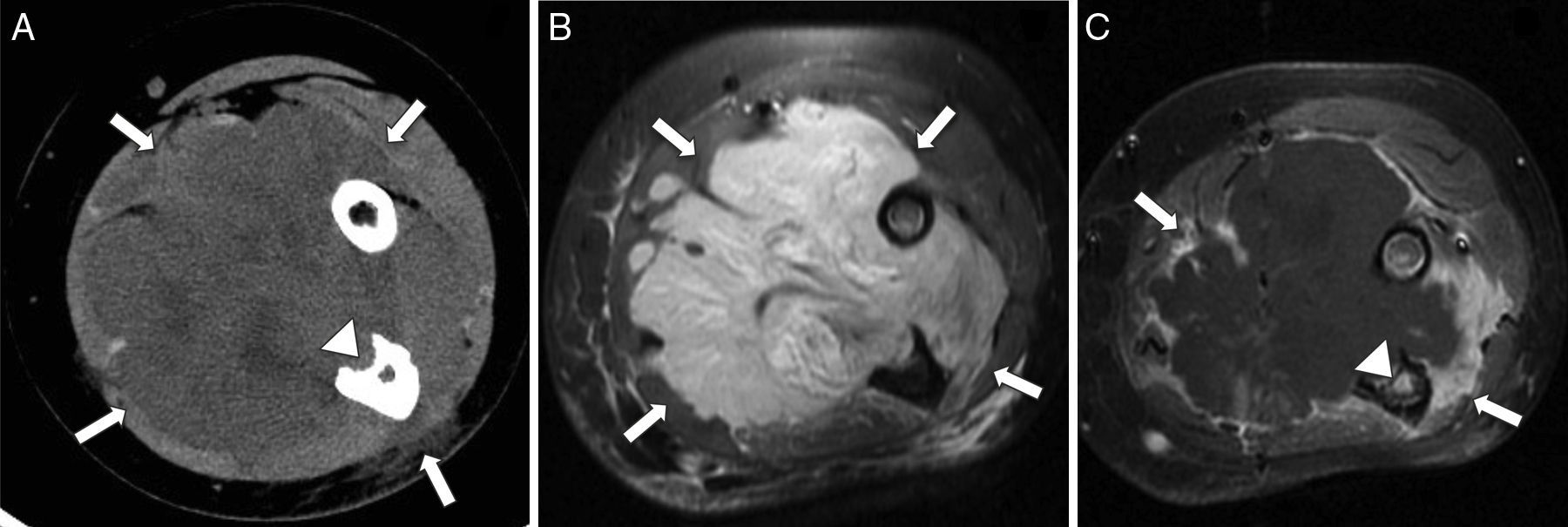
El RMS de las extremidades es casi siempre del subtipo alveolar, que representa alrededor del 50% de los casos4–6. Se presenta típicamente como una masa no dolorosa, aunque suele ser detectado más rápido por los propios pacientes y los pediatras como un bultoma que puede o no estar acompañado de edema, dolor y eritema; algunos pacientes tienen adenopatías en el momento del diagnóstico y diseminación a través de las fascias19. El diagnóstico diferencial debe hacerse con el fibrosarcoma infantil, el sarcoma sinovial, el sarcoma de Ewing extraesquelético y tumores vasculares, entre otros (fig. 9).
 Corte transversal de TC con contraste intravenoso que muestra un aumento patológico de volumen no delimitable de la musculatura (flechas), con infiltración y destrucción ósea del cúbito (punta de flecha). B) Corte transversal de RM T2-FS que confirma la presencia de una extensa lesión hiperintensa (flechas) que engloba los distintos grupos musculares del antebrazo. C) Corte transversal de RM T1-FS poscontraste que muestra realce periférico de la tumoración (flechas) y un foco de captación intramedular en el cúbito (punta de flecha), que confirma la infiltración de la medular.")
Rabdomiosarcoma alveolar de antebrazo. A) Corte transversal de TC con contraste intravenoso que muestra un aumento patológico de volumen no delimitable de la musculatura (flechas), con infiltración y destrucción ósea del cúbito (punta de flecha). B) Corte transversal de RM T2-FS que confirma la presencia de una extensa lesión hiperintensa (flechas) que engloba los distintos grupos musculares del antebrazo. C) Corte transversal de RM T1-FS poscontraste que muestra realce periférico de la tumoración (flechas) y un foco de captación intramedular en el cúbito (punta de flecha), que confirma la infiltración de la medular.
Alrededor del 10% de los RMS se encuentran en el tronco4 y existen algunos casos de tumores originados sobre malformaciones adenomatoides quísticas5. Pese a que el RMS es el tumor maligno más frecuente de la vía biliar, es excepcional y su origen en el tracto biliar suele ser difícil de comprobar5,20.
Los RMS primarios de localización intratorácica, intraperitoneal o pélvica suelen ser muy grandes en el momento del diagnóstico, lo que dificulta una escisión quirúrgica completa, en parte debido a la infiltración de los grandes vasos (fig. 10).
 Corte transversal de TC abdominal con contraste intravenoso que muestra una extensa tumoración retroperitoneal (flechas), de densidad heterogénea, que desplaza y comprime las vísceras abdominales. B) Corte transversal de RM potenciada en T2: la tumoración es hiperintensa, con un área de degeneración necrótico-quística (asterisco) en su interior, que engloba a los vasos retroperitoneales (flechas negras) y el estómago (flecha blanca).")
Rabdomiosarcoma alveolar retroperitoneal. A) Corte transversal de TC abdominal con contraste intravenoso que muestra una extensa tumoración retroperitoneal (flechas), de densidad heterogénea, que desplaza y comprime las vísceras abdominales. B) Corte transversal de RM potenciada en T2: la tumoración es hiperintensa, con un área de degeneración necrótico-quística (asterisco) en su interior, que engloba a los vasos retroperitoneales (flechas negras) y el estómago (flecha blanca).
El tratamiento requiere un abordaje multidisciplinario que incluya quimioterapia, cirugía y radioterapia.
El tratamiento quirúrgico comienza con la toma de la biopsia, que debe ser realizada por un equipo entrenado en el tratamiento de esta patología. Una vez confirmado el diagnóstico anatomopatológico, se realiza la estadificación inicial, se administra quimioterapia para reducir el tamaño de la masa y se valora la respuesta al tratamiento. Posteriormente se realiza la intervención quirúrgica, tras la cual se hace una nueva reestadificación posquirúrgica basándose en la resección completa o no y en la presencia de enfermedad residual según los criterios del Intergroup Rhabdomyosarcoma Study (IRS-IV). Los tumores paratesticulares son el único caso en que está indicada la biopsia escisional de inicio5.
La radioterapia se administra en casi todos los pacientes después de la cirugía, excepto en aquellos sin evidencia de enfermedad microscópica tras la extirpación tumoral, en los que debe realizarse quimioterapia prolongada.
Este tratamiento multidisciplinario ha producido un incremento progresivo de la tasa de curación, que alcanzaba un porcentaje cercano al 70% en 199121, con unas tasas de supervivencia libre de enfermedad a los 3 años del 86%, el 80%, el 68% y el 25% para los pacientes en los estadios I, II, III y IV, respectivamente6,12.
El pronóstico del RMS se relaciona con la histología, la edad del paciente, el lugar de origen, el diámetro mayor del tumor, la resecabilidad, la afectación de ganglios linfáticos y la administración de radioterapia en algunos casos específicos, así como con las características biológicas distintivas de las células tumorales. Para su determinación, se utiliza un sistema de estratificación creado por el EpSSG (tabla 2).
Factores pronósticos del EpSSG (European pediatric Soft-tissue sarcoma Study Group)
| Favorable | Desfavorable | |
|---|---|---|
| Histología | Embrionario | Alveolar |
| Estadio IRS poscirugía | A mayor grado, más desfavorable | |
| Sitio del tumor | Cabeza y cuello no parameníngeo Órbita Genitourinario no vejiga ni próstata | |
| Adenopatías | N0 | N1 |
| Tamaño tumoral | ≤5cm | >5cm |
| Edad | <10 años | 10 años o más |
Adaptada de Van Rijn et al.5
Aproximadamente un 30% de los pacientes recae, y de ellos, un 50-95% fallece por progresión de la enfermedad6.
ConclusionesEl RMS es un tumor agresivo, con clínica inespecífica y pronóstico variable en función de su localización, subtipo histológico y estadio. De las variantes pediátricas, el subtipo embrionario es el más frecuente y se localiza preferentemente en la cabeza, el cuello y el tracto genitourinario. El subtipo alveolar es la forma más agresiva, con frecuencia se localiza en el tronco y las extremidades, y se caracteriza por un aspecto histológico único y por poseer una anomalía genética específica entre los genes PAX3 y PAX7 con el gen forkhead.
Pese a que los estudios de imagen no tienen características definitorias para su diagnóstico, son esenciales para la estadificación y el abordaje terapéutico, así como en la planificación quirúrgica y la valoración de la respuesta tumoral al tratamiento.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que en este artículo no aparecen datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores declaran que en este artículo no aparecen datos de pacientes.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Autoría- 1
Responsables de la integridad del trabajo: SMCR y EJIC.
- 2
Concepción del trabajo: SMCR y EJIC.
- 3
Diseño del estudio: no aplica.
- 4
Obtención de los datos: no aplica.
- 5
Análisis e interpretación de los datos: no aplica.
- 6
Tratamiento estadístico: no aplica.
- 7
Búsqueda bibliográfica: SMCR y EJIC.
- 8
Redacción del trabajo: SMCR.
- 9
Revisión crítica del manuscrito con aportaciones intelectualmente relevantes: EJIC.
- 10
Aprobación de la versión final: SMCR y EJIC.

