




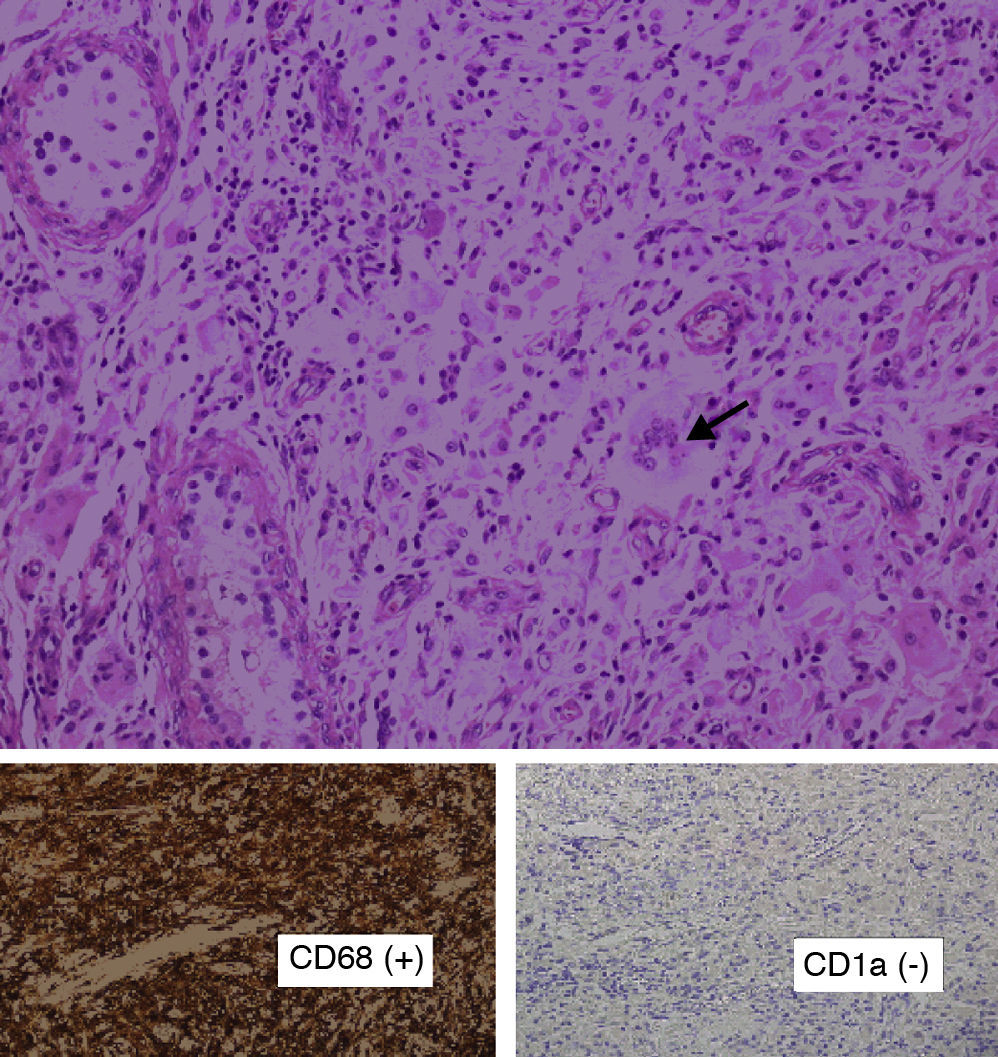
La enfermedad de Erdheim-Chester (EC) es una histiocitosis de células no-Langerhans que cursa con infiltración xantogranulomatosa multiorgánica por histiocitos CD68+/CD1a-. Se recogen las principales características de 12 pacientes diagnosticados de esta rara enfermedad.
Pacientes y métodoSe revisaron las historias clínicas y los hallazgos anatomopatológicos de 12 casos diagnosticados de enfermedad de EC en 7 hospitales terciarios de la península. Se consideró el diagnóstico de esta enfermedad ante un cuadro clínico compatible e infiltración tisular por histiocitos CD68+/CD1a-.
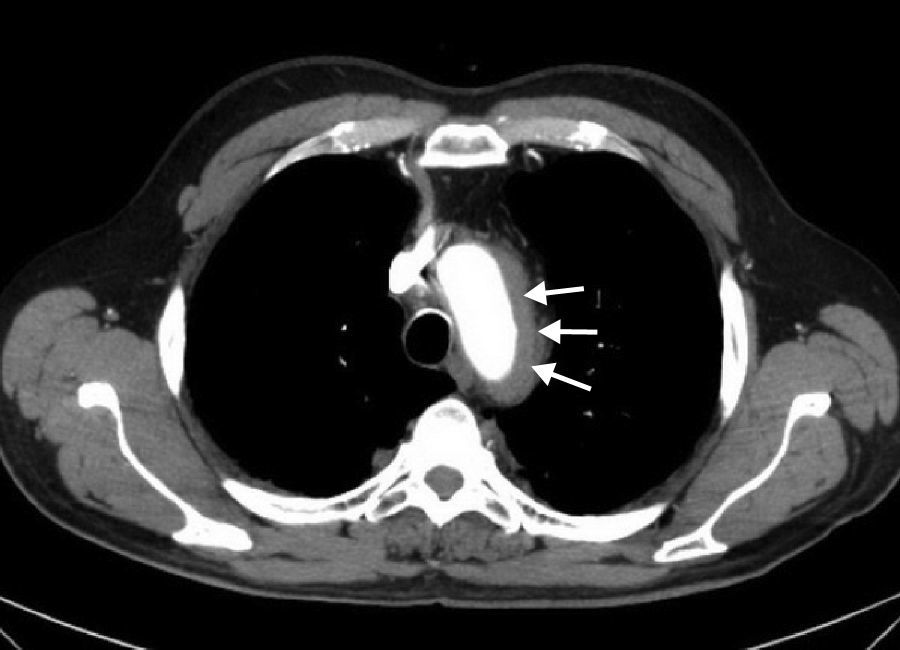
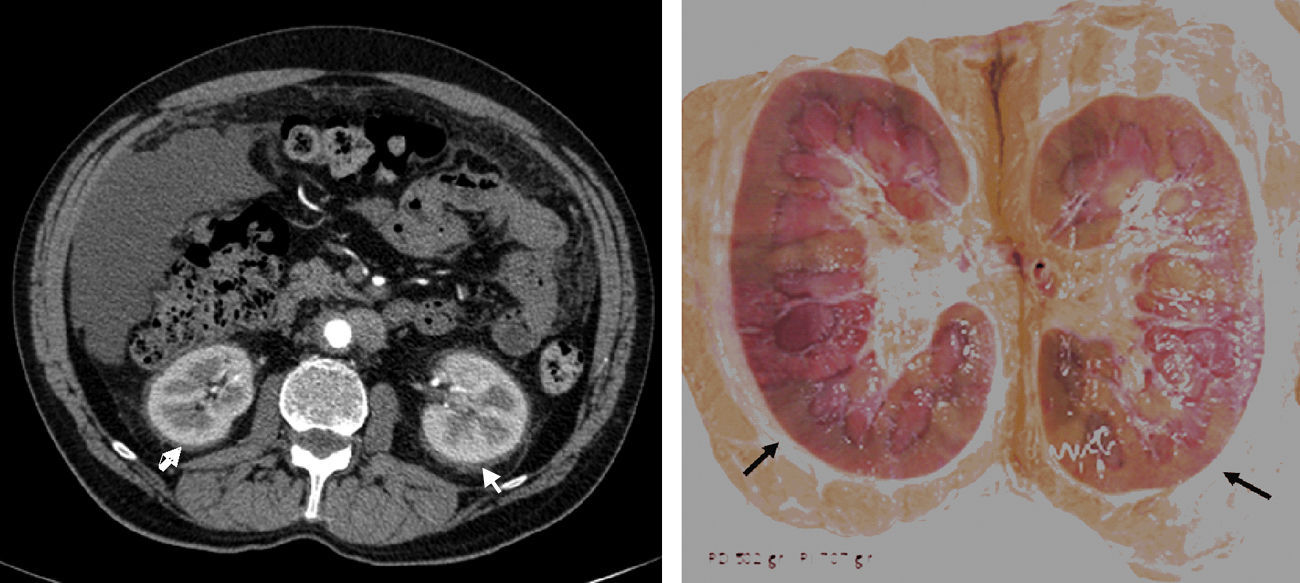
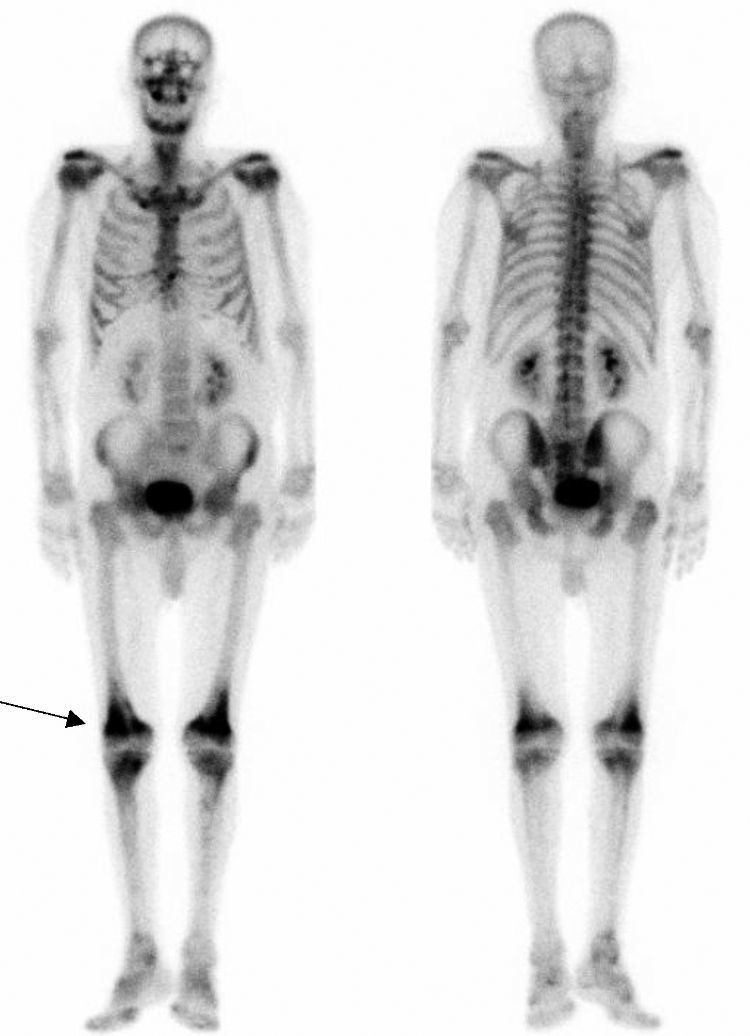
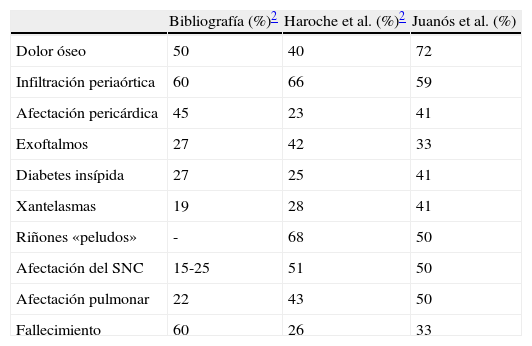
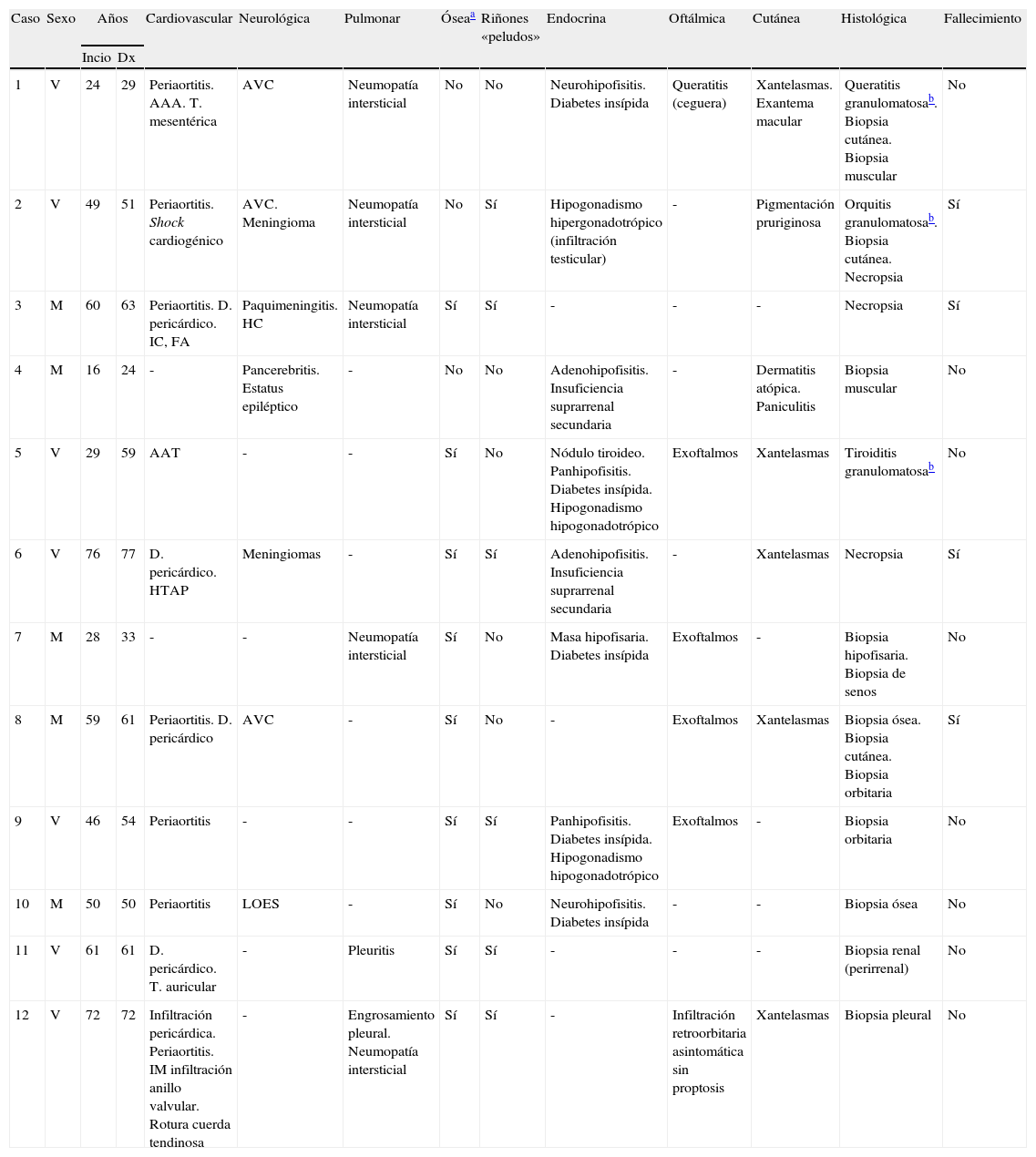
ResultadosSe incluyó en el estudio a 12 pacientes, 7 varones, con una mediana de seguimiento de 36 meses (rango IQ: 20-84). La mediana de edad al inicio clínico de la enfermedad y en el momento del diagnóstico histológico fue de 49 (rango IQ: 28-61) y 56 años (37-62), respectivamente. En 6 casos se realizaron múltiples biopsias para poder llegar al diagnóstico, mientras que en 3 fue la revisión de las mismas piezas anatomopatológicas en un adecuado contexto de sospecha clínica lo que permitió el diagnóstico. Las manifestaciones neurológicas presentaron una asociación estadísticamente significativa con la mortalidad (p<0,05). La característica afectación ósea en forma de osteoesclerosis metadiafisaria de huesos largos se detectó en 9 casos.
ConclusionesLa enfermedad de EC presenta una gran heterogeneidad en sus manifestaciones clínicas. Es preciso un alto índice de sospecha y una estrecha colaboración entre clínicos y patólogos para llegar al diagnóstico de esta enfermedad.
Erdheim-Chester disease (EC) is a rare form of non-Langerhans’ cell histiocytosis. It is characterized by the xanthomatous infiltration of tissues with foamy CD68+/CD1a- histiocytes. We report a series of 12 patients diagnosed with EC.
Patients and methodsWe reviewed the clinical, pathological and therapeutic aspects of 12 cases diagnosed with EC at 7 tertiary teaching hospitals in Spain. Patients were included if tissue infiltration by histiocytes CD68+/CD1a- could be demonstrated in an appropriate clinical setting.
ResultsTwelve patients (7 male) were included. Median follow-up was 36 months (IQR: 20-84). The median age at the time of clinical onset and pathological diagnosis was 49 (IQR: 28-61) and 56 years (IQR: 37-62), respectively. In 6 cases multiples biopsies were performed (skin, muscle, testicular) previous to diagnosis, which was confirmed in 3 cases after a carefully review of pathological tissues. Neurological involvement was independently associated with mortality (P<.05). Characteristic long bone osteosclerosis was detected in 9 patients.
ConclusionEC is a multisystemic and heterogeneous clinicopathological condition. A high index of suspicion and fluent communication between clinicians and pathologists is necessary to achieve a correct diagnosis.
Artículo

Comprando el artículo el PDF del mismo podrá ser descargado
Precio 19,34 €
Comprar ahora
