





Development of mesoporous silica nanoparticles as carriers for drug delivery systems has increased exponentially during the last decade. The present work is focused on the synthesis of silica carriers by sol–gel from tetraethyl orthosilicate (TEOS) as precursor of silica and cetyltrimethylammonium bromide (CTAB) as pore generating agent. The synthesis conditions were modified varying the molar ratio of water/TEOS, NH3/TEOS and amount of CTAB. The silica particles were characterized by scan electron microscopy techniques (FESEM), high resolution transmission electron microscopy (HR-TEM), N2 adsorption–desorption isotherms, Zeta-potential and Dynamic Light Scattering (DLS). The results show that the specific surface area and the porosity of silica particles were strongly affected by the addition of CTAB and the amount of H2O. The dispersion and stability of silica mesoporous particles is achieved in spite of the high surface reactivity. The synthesis formulation affects considerably to the particle morphology, which changes from spheres to rods when the molar ratio of H2O increases. A maximum specific surface area of 1480m2/g was obtained with pore sizes ranging 2.5–2.8nm.
El interés por el uso de partículas mesoporosas de sílice como vehículos para sistemas de liberación de fármacos ha aumentado exponencialmente en la última década. Este trabajo se centra en la síntesis de portadores de sílice por sol-gel usando tetraetilortosilicato (TEOS) como precursor de sílice y bromuro de cetiltrimetilamonio (CTAB) como agente generador de poros. Se ha analizado el efecto en las propiedades morfológicas de los cargos de sílice de la modificación de algunas condiciones de síntesis como las relaciones molares agua/TEOS y amoniaco/TEOS, o la cantidad de CTAB. Las partículas obtenidas se han caracterizado mediante microscopia electrónica de barrido y de emisión de campo, por microscopia electrónica de transmisión, adsorción/desorción de N2, potencial zeta y dispersión dinámica de luz (DLS). Los resultados muestran que la superficie específica y la porosidad de las partículas de sílice se ven especialmente afectadas por la cantidad de CTAB, alcanzándose superficies específicas de 1.480 m2/g con una porosidad media de 2,5-2,8nm. Las partículas mesoporosas de sílice permanecen dispersas y estables a pesar de su elevada reactividad superficial. La formulación de la disolución precursora cambia considerablemente la morfología de las partículas, que pasan de ser esféricas a adoptar una morfología de varillas cuando aumenta la cantidad de H2O adicionada.
In the last decades, drug delivery technologies have been changed substantially. In general, the human body is a complex system which depends on the disease to be treated, and hence, the procedure to introduce each drug in the tissue is different. Moreover, the secondary effect of drugs is that can affect to other parts of the body, so drugs must only interact with the target and, at the same time, its release should be controlled to avoid the secondary effects. Amount carriers, mesoporous materials have been emerged as novel tools in biomedical applications [1,2]. Mesoporous structures are characterized by having a large specific surface area and pores with diameters between 2 and 50nm. Among inorganic structures, silica (SiO2) materials have been intensely studied as potential applications in catalysis, biological, biomedicine, etc., due to its outstanding characteristics including availability for mass production and simple synthesis method [3,4]. Mesoporous silica nanoparticles (MSNs) can be used as host materials for transporting therapeutics medicaments or encapsulation of molecules [5,6]. Good biocompatibility, high loading capacity, the possibility of attachment target ligands for specific cellular recognition or the design of well-defined and tunable porosity, make MSNs suitable for drug delivery [7,8]. But for all applications, the morphology of the material is one of the most important aspects. One common synthesis route to obtain MSNs is based on the use of templating agents, typically a surfactant which neutral or charged and that acts as structure-directing agent [9]. In fact, first mesoporous silica particles were produced via modification of the Stöber process by using soft template strategy [10]. In general, MSNs are synthesized by using a silica precursor (tetraethylorthosilicate or sodium silicate) in an alcoholic solution under basic conditions and incorporating a surfactant [11,12]. Some authors have reported the mechanism to obtain silica nanospheres and nanorods changing the surfactant concentration [13,14] and sol dilution [15]. The interaction between the surfactant with the Si–O–Si species was studied, and the pore sizes, shapes and order, as well as MSNs morphology, were related with the characteristics of the surfactant (size, length, etc.) and the formation of the micelles [16,17]. The final morphology of the particles affect to the capacity of absorption of drug and their subsequent release [18].
On the other hand, synthesis of mesoporous silica particles in non-alcoholic medium were described in the literature [14,15] but the formation of spherical particles is limited by the amount of the surfactant (<0.8–1wt.%). The specific surface area of the mesoporous silica particles reported is 1030–1070m2/g with a pore volume of 0.81–0.85cm3/g. Wang et al. [13] considered the use of EtOH but change the amount of water for a fixed amount of CTAB (4.1wt.%), obtaining spherical and rod-like particles with an ordered mesoporous structure. Silica particles reached a specific surface area of 1500m2/g and a pore volume of 0.86cm2/g. the dilution of the sol changes the specific surface area and transforms the morphology of the particles form spherical to rod-like, evidencing that both the evolution of the mesoporosity and the morphology of silica particles depend on the micelles formation and ordering.
The aim of the work is to study the particle size morphology and dispersion of mesoporous silica nanoparticles prepared by sol–gel by changing simultaneously the water content and amount of surfactant (CTAB). The silica nanoparticles were characterized by field-emission scanning (FESEM) electron microscopy, high resolution transmission electron microscopy (HR-TEM), zeta-potential measurements and Dynamic Light Scattering, and N2 adsorption–desorption measurements. Difference morphologies, spherical and rod-like, have been obtained and compared considering all of synthesis parameters.
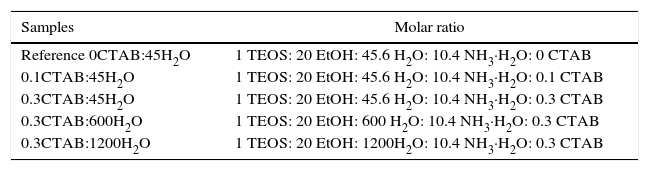
ExperimentalAlkoxides precursor used was tetraethyl orthosilicate, purchased in ABCR (TEOS, 99%) and cetyltrimethylammonium bromide (CTAB, 99%) as surfactant, from Aldrich. The rest of precursors used were analytical grade reactant from Aldrich. Silica sol was prepared by mixing EtOH, H2O and ammonia solution (NH4OH). Afterwards, CTAB was added to the first solution and maintained under stirring for 15min. Then, TEOS was added drop by drop under continuous stirring for 2h at room temperature. The solution turned opaque almost immediately, indicating that the reaction has started. The white powder precipitated was filtrated and washed with deionized water. The particles were dried at room temperature overnight. Then, the particles were calcined at 550°C for 3h to remove the surfactant. The final molar ratio of TEOS/EtOH was fixed to 1/20 and the molar ratios of H2O/NH3·H2O/CTAB were varied, Table 1.
SiO2 sol–gel formulations.
| Samples | Molar ratio |
|---|---|
| Reference 0CTAB:45H2O | 1 TEOS: 20 EtOH: 45.6 H2O: 10.4 NH3·H2O: 0 CTAB |
| 0.1CTAB:45H2O | 1 TEOS: 20 EtOH: 45.6 H2O: 10.4 NH3·H2O: 0.1 CTAB |
| 0.3CTAB:45H2O | 1 TEOS: 20 EtOH: 45.6 H2O: 10.4 NH3·H2O: 0.3 CTAB |
| 0.3CTAB:600H2O | 1 TEOS: 20 EtOH: 600 H2O: 10.4 NH3·H2O: 0.3 CTAB |
| 0.3CTAB:1200H2O | 1 TEOS: 20 EtOH: 1200H2O: 10.4 NH3·H2O: 0.3 CTAB |
A reference sol was prepared following the same process but without surfactant addition (labelled as Reference 0CTAB:45H2O). The molar ration of water/TEOS was varied between 45 and 1200, while the CTAB molar ratio was fixed at 0, 0.1 and 0.3 (0, 1.5 and 4.1wt.%).
The particle size and morphology were examined by Field Emission Scanning Electron Microscopy (FESEM) using S-4700 microscope (Hitachi, Japan). High-Resolution Transmission Scanning Microscope (HR-TEM) was used to confirm the homogeneity and porous structure of the films. The equipment used was a JEM-2011 (JEOL Ltd., Japan) working at 200KV.
A ZetasizerNano ZS (Malvern, UK) was used to determine the zeta potential and particle size distribution of the mesoporous silica nanoparticles using Dynamic Light Scattering (DLS). Suspensions were prepared with concentrations of 0.1–0.01g/L using 10−2M KCl as solvent and inert electrolyte, to maintain the ionic strength of the medium. pH adjustments of the suspensions was carried out by addition of small quantities of 0.1M HNO3 or TMAH and controlled with a pH probe (Metrohm AG, Germany). Subsequently, homogenization was achieved by sonication, using a UP400S Ultrasonication probe (Hielscher, Germany) for an optimized period of time of 30s.
Finally, textural properties were analyzed by means the nitrogen adsorption isotherm, using a Micromeritics ASAP 2020 porosimeter. The samples were previously degassed at 250°C for 20h. The Brunauer–Emmet–Teller method was applied to calculate the specific surface area (SBET) and pore size from the desorption curve. The average pore diameter was calculated from the pore volume assuming cylindrical pore and SBET calculated by BET surface area.
Results and discussionIn the present study, mesoporous silica particles of different morphology and particle size were synthesized by varying the amount of surfactant and H2O (Table 1). Fig. 1 shows the FESEM images of mesoporous silica particles prepared by sol–gel as a function of the surfactant and water amount. The micrographs show a variety of morphology and sizes. The reference particle (0CTAB:45H2O sample) exhibits a perfect and homogeneous spherical shape with an external surface very smooth and with an average diameter of 740nm. On the other hand, silica particles prepared at molar ratio of water/TEOS=45 but changing the amount of CTAB show a wide particle size distribution, ranging from 150 to 750nm (Fig. 1b and c). More homogeneous spherical particles are obtained for low concentration of CTAB. So, the CTAB concentration affect to the growth of particles as were also reported to B. Chen [15] and G. Lelong [14]. Thereby, in order to analyze in more detail the silica particles growth, 0.1CTAB:45H2O and 0.3CTAB:45H2O samples was observed by HR-TEM (Fig. 2). Micrographs demonstrated a variety of particle shapes and sizes. For low concentration of CTAB agglomerated silica particles are observed with a population heterogeneous in size (Fig. 2a and c). However, for 0.3CTAB:45H2O, the silica particles are not separated individually, so “Ostwald Ripening” growth can be presumed. Fig. 2c shows a transformation of the particle morphology from spherical to an ellipsoidal particle that can evolve to an elongated rod. Different interactions between alkoxysilane and CTAB at the micelle/water interface could contribute to the drastic changes in growth of the particles. The mesopores could start to grow along the long axes and give rise to small ellipsoidal particles. On the other hand, at high magnification, HR-TEM confirm the presence of mesoporous in all the samples (Fig. 2b and d), with randomly oriented pore structure.
 0CTAB:45H2O, (b) 0.1CTAB:45H2O, (c) 0.3CTAB:45H2O, (d) 0.3CTAB:600H2O and (e) 0.3CTAB:1200H2O.")
 and of 0.3CTAB:45H2O sample (c and d).")
The variation in the morphology of the particles is more accentuated with changing the water amount (Fig. 1d and e). The particle morphology changes completely and rod-like particles were obtained. In fact, the length of rods depends on the water content. The average particle size of 0.3CTAB:600H2O is larger than that of 0.3CTAB:1200H2O, being 1.6μm and 530nm, respectively. It means that the growth of the particles is promoted in a certain direction. The textural morphology is associated with the surfactant and water concentration because of the mechanisms of micelle formation and arrangement of CTAB are influenced by the precursor formulation. By increasing the amount of water the silica particles elongates and grows following the micelle configuration. The solution dilution changes the configuration of the surfactant micelles forming aggregates that encapsulate the silica precursor decreasing the hydrolysis of the TEOS and promoting the growth of the mesoporous silica particle in the direction that is perpendicular to the pore-alignment [14].
Zeta potential value of particles in suspension is defined as the potential at the shear layer formed in the double layer of the surface of the particle when it is immersed in a liquid media. Zeta-potential measurements are crucial to prepare stable and dispersed suspensions of mesoporous particles in water and to validate the experimental conditions for the DLS measurement. If all the particles in suspension exhibit a large positive or negative zeta potential, the agglomeration of the particles will be prevented. A common separating line between unstable and stable suspensions is taken as +30 or −30mV; particles having zeta potentials beyond these limits are generally considered as electrostatically stable. In our case, all the samples have absolute value of zeta-potential around 40–45mV (graphs not shown), which indicates that the post-synthesis suspensions are stable. This zeta potential is higher than that reported for other authors (−30.7mV)[19]. On the other hand, the zeta-potential was negative, evidencing the silica nature of the particles. Only, the suspension of 0.3CTAB:1200H2O was not stable, observing a small sediment layer at the bottom of the cell, so it was not analyzed. The tendency to form agglomerates could be associated to the high reactivity of the particles surface.
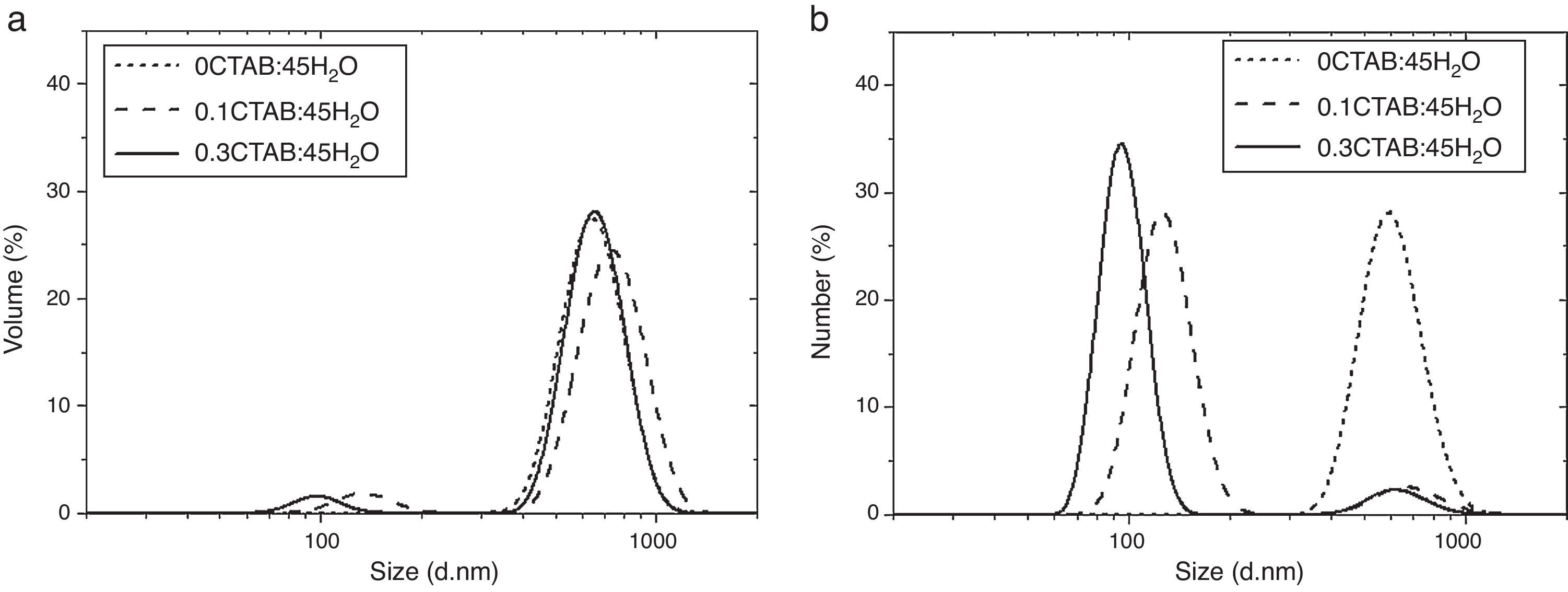
After zeta potential, DLS measurements were performed to determine the particle size and to evaluate the influence of the CTAB content in the particle size. Fig. 3 and Table 2 shows and summarizes the DLS and FESEM observations for the samples prepared with two different contents of CTAB and without CTAB, with molar ratio TEOS/H2O=1/45 and 1/600, respectively. When no surfactant was added, the population of particles is represented by a monomodal distribution. There is only one width peak, centred on 600nm, and mean diameters in volume and number (Dv50 and Dn50) are quite similar. Despite of the size distribution ranges from ∼300nm to ∼1000nm, fine and coarse fractions in the dispersion are almost negligible. This fact is supported by the low value of the Polydispersity Index (PdI), which strengthen the idea of the synthesis of well dispersed population of spherical particles.
 volume (Dv) and (b) number (Dn) for 0CTAB:45H2O, 0.1CTAB:45H2O and 0.3CTAB:45H2O.")
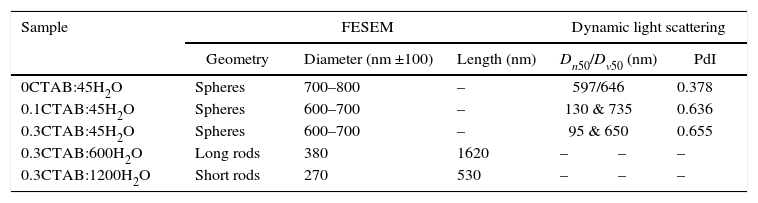
Particle size measured by FESEM, and main particle size and polydispersity index determined for DLS for 0CTAB:45H2O, 0.1CTAB:45H2O, 0.3CTAB:45H2O, 0.3CTAB:600H2O and 0.3CTAB:1200H2O samples.
| Sample | FESEM | Dynamic light scattering | ||||
|---|---|---|---|---|---|---|
| Geometry | Diameter (nm ±100) | Length (nm) | Dn50/Dv50 (nm) | PdI | ||
| 0CTAB:45H2O | Spheres | 700–800 | – | 597/646 | 0.378 | |
| 0.1CTAB:45H2O | Spheres | 600–700 | – | 130 & 735 | 0.636 | |
| 0.3CTAB:45H2O | Spheres | 600–700 | – | 95 & 650 | 0.655 | |
| 0.3CTAB:600H2O | Long rods | 380 | 1620 | – | – | – |
| 0.3CTAB:1200H2O | Short rods | 270 | 530 | – | – | – |
When surfactant is added to the synthesis, double peaks appear in the particle size distributions. The DLS measurements evidence, as the FESEM micrographs (Fig. 1b and c), the presence of a wider population of particles with different sizes for 0.1CTAB:45H2O and 0.3CTAB:45H2O samples, point out by the PdI increases. For both samples, the coarse particle populations are relevant in volume while nanoparticles are more representative in number. This fact explains the divergence between the plots of volume and number distributions. Dv50 and Dn50 are similar for the micro and nano-populations. The distributions of the coarse particles match the monomodal distribution of the reference material, without CTAB (Dv50=735–650nm), while the fine fraction slightly decreases in size with the amount of CTAB, size turning from 130 to 95nm. The samples 0.3CTAB-600 H2O and 0.3CTAB-1200 H2O were not analyzed by DLS due to their rods-like morphologies produced erroneous and confused distributions.
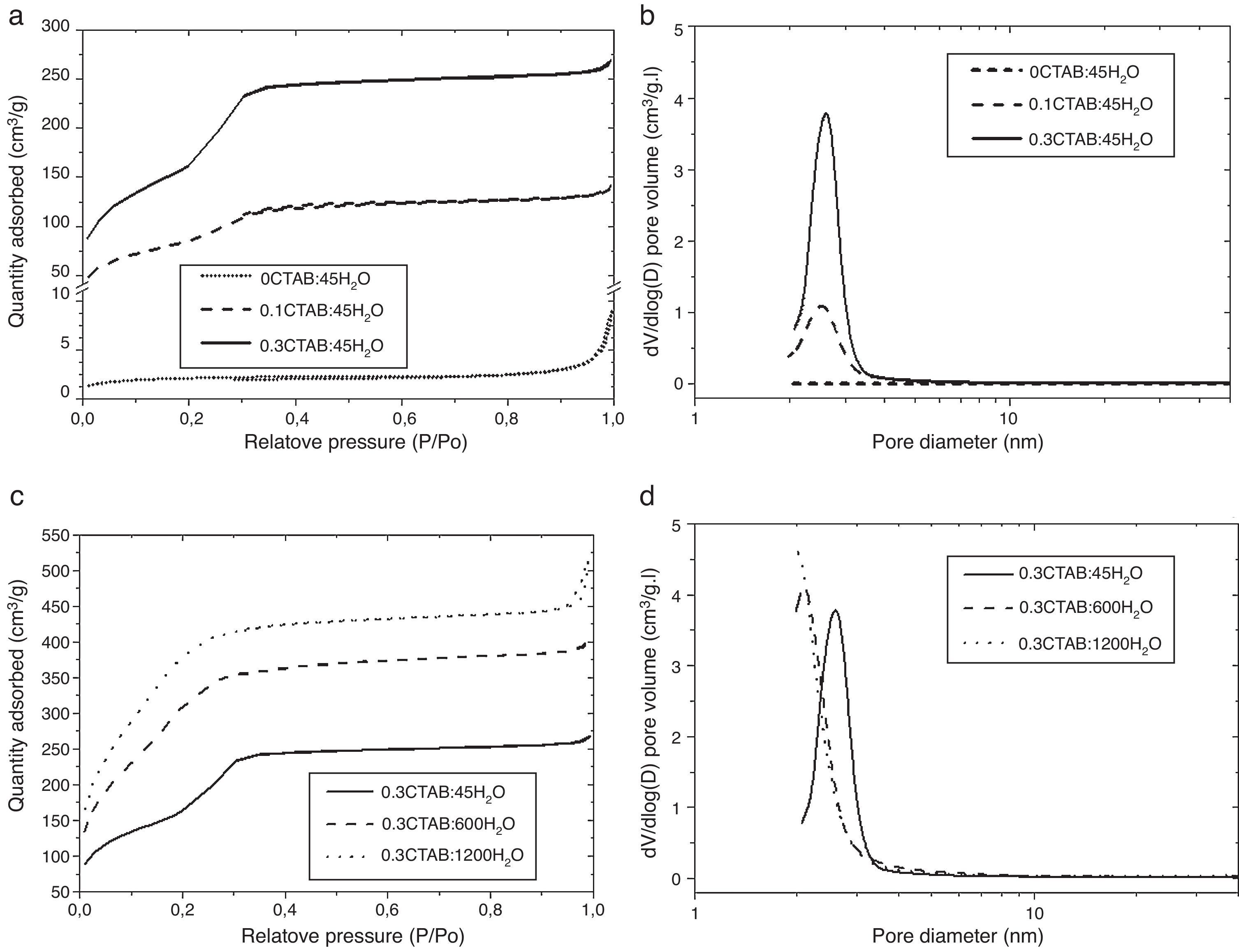
Then, porosity and accessibility to the pores were studied by N2 adsorption/desorption measurements. Fig. 4 shows the adsorption isotherm (a) and pore size distribution (b) for 0CTAB:45H2O, 0.1CTAB:45H2O, 0.3CTAB:45H2O samples. Similarly plots in Fig. 4(c) and (d) shows the adsorption isotherm and pore size distribution, for 0.3CTAB:600H2O and 0.3CTAB:1200H2O samples, respectively.
 Adsorption isotherms for the samples 0CTAB:45H2O, 0.1CTAB:45H2O, and 0.3CTAB:45H2O and (c) for the samples 0.3CTAB:45H2O, 0.3CTAB:600H2O and 0.3CTAB:1200H2O. Curves (b) and (d) are the pore size distribution corresponding to the same samples described previously in (a) and (c) respectively.")
(a) Adsorption isotherms for the samples 0CTAB:45H2O, 0.1CTAB:45H2O, and 0.3CTAB:45H2O and (c) for the samples 0.3CTAB:45H2O, 0.3CTAB:600H2O and 0.3CTAB:1200H2O. Curves (b) and (d) are the pore size distribution corresponding to the same samples described previously in (a) and (c) respectively.
It can be observed that the isotherms for samples with and without surfactant are different. The isotherm of 0CTAB:45H2O sample shows a non-porous material. The addition of surfactant changes completely the type of isotherm. For 0.1CTAB:45H2O sample shows a Type IV isotherm, where an increase in adsorption take place above P/P0=0.2, suggesting the capillary condensation of N2 within pore and indicating the presence of mesopores. Then, the isotherm is relatively linear. In the case of 0.3CTAB:45H2O sample, high adsorption is observed at very low relative pressure. As in the previous case, a large presence of mesopores, characteristic of a narrow pore size distribution, is observed. But the sharp increment of volume occurs at the same P/P0 (0.2) which is associated with the lower pore size of this composition comparing with the 0.1CTAB:45H2O sample. It means that not just specific surface area have influence but also the pore size. On the other hand, the isotherms for the samples with high amount of water are different. The most important result is the higher amount of N2 adsorbed for 0.3CTAB:600H2O and 0.3CTAB:1200H2O samples compared to 0.3CTAB:45H2O sample, associated with an increment of the specific surface area. The isotherms for 0.3CTAB:600H2O and 0.3CTAB:1200H2O have a similar absorbance, so it is reasonable to assume that they have similar pores in the range of mesoporous (2 or 3nm). On the other hand, although the adsorption of N2 starts at relatively low pressures the complete filling of the pores occurs at the same pressure (P/P0=0.3).
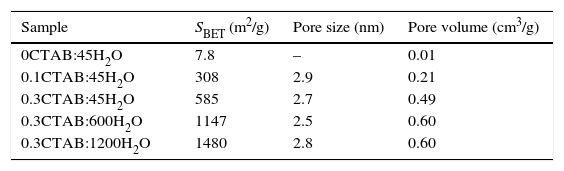
Table 3 summarize the porosity analysis of all the samples. The specific surface area (SBET) increases with the CTAB content from 308 to 585m2/g. The addition of CTAB enhances in two orders of magnitude the specific surface area, leading to an increment of the pore volume of 0.21–0.49cm3/g. The diameter of pores appears unaffected (∼2.5 to 2.8nm) for both samples. This pore size could be tailored selecting different type of surfactants. It is important to point out that the higher is the amount of CTAB, the higher is the volume of N2 adsorbed onto the solid, while pore size is maintained in the same range. Consequently, the addition of CTAB produces samples with more quantity of interconnected pores, and thus, the specific surface area increases. For the higher concentration of CTAB, the corresponding amount of silica precursor per surfactant molecule falls down and, the condensation of TEOS around the micelles formed by the organic molecules leads to a decrease in the thickness of the silica pore walls, as well as, the increase of the mesoporosity of the particles. Moreover, the thin silica pore walls could affect to the morphology and size of the particles. In addition, the increment of water promotes a large increase of the specific surface area above 1480m2/g. As in the other case, the pore volume increases while the sizes are keeping constant.
Specific surface area, pore size and pore volume for 0CTAB:45H2O, 0.1CTAB:45H2O, 0.3CTAB:45H2O, 0.3CTAB:600H2O and 0.3CTAB:1200H2O samples.
| Sample | SBET (m2/g) | Pore size (nm) | Pore volume (cm3/g) |
|---|---|---|---|
| 0CTAB:45H2O | 7.8 | – | 0.01 |
| 0.1CTAB:45H2O | 308 | 2.9 | 0.21 |
| 0.3CTAB:45H2O | 585 | 2.7 | 0.49 |
| 0.3CTAB:600H2O | 1147 | 2.5 | 0.60 |
| 0.3CTAB:1200H2O | 1480 | 2.8 | 0.60 |
The mesoporous silica particles obtained with different morphology and with high specific area could be attractive candidates for using as drug deliverable container in future works.
ConclusionsMesoporous silica nanoparticles (MSNs) have been prepared by sol–gel process, using tetraethyl orthosilicate (TEOS) as alkoxide precursor and cetyltrimethylammonium bromide (CTAB) as surfactant.
The surfactant content plays an important effect in the particle morphology changing from dispersed nanospheres to agglomerates, changing the hydrolysis of TEOS and the micellization of CTAB. As expected, the pore size does not change and pore diameters between 2.5 and 2.8nm are obtained.
The addition of CTAB produces samples with high specific surface area (585m2/g) and pore volume (0.49cm3/g), for silica nanocarriers with a main particle size in volume 600–700nm. In general, the addition of CTAB induces silica mesoporous structure growth following an Ostwald ripening mechanism as HR-TEM images evidenced.
On the other hand, the dilution of sol leads to the formation of rod-like particles with a specific surface area up to 1480m2/g. However those particles present a high surface reactivity that does not allow their dispersion in water.
In general, a wide range of configurations of particles depending on processing conditions can be obtained and could be used as drug deliverable container.
The authors acknowledge the support of the project S2013/MIT-2862 and MAT2015-70780-C4-1 and Dr. Z Gonzalez acknowledges to MINECO through the grant PTQ-13-05985. To Jesus Canales Vázquez from the Castilla la Mancha University for his support in HR-TEM analysis.

