




Carbonated hydroxyapatite (CHAp) and β-tricalcium phosphate (β-TCP) have been employed for decades as constituents of scaffolds for bone regeneration because they chemically resemble bone mineral. In this study, the feasibility to manufacture CHAp/β-TCP scaffolds by pseudomorphic transformation of casted blocks of gypsum was investigated.
The transformation was carried out by immersing the precursor gypsum block in 1 M (NH4)2HPO4/1.33 M NH4OH solution with liquid/solid ratio of 10 mL/g and autoclaving at 120 °C and 203 kPa (2 atm) for 3 h at least. Neither shape nor dimensions significantly changed during transformation. The composition of scaffolds treated for 3 h was 70 wt.% CHAp and 30 wt.% β-TCP, and their compressive and diametral compressive strengths were 6.5 ± 0.7 and 5.3 ± 0.7 MPa, respectively. By increasing the time of treatment to 6 h, the composition of the scaffold enriched in β-TCP (60 wt.% CHAp and 40 wt.% β-TCP) but its compressive and diametral compressive strengths were not significantly affected (6.7 ± 0.9 and 5.4 ± 0.6 MPa, respectively).
On the basis of the results obtained, it was concluded that this route is a good approach to the manufacturing of biphasic (CHAp/β-TCP) scaffolds from previously shaped pieces of gypsum.
La hidroxiapatita carbonatada (CHAp) y el β-fosfato tricálcico (β-TCP) han sido utilizados durante décadas como constituyentes de los andamios para regeneración ósea, debido a su semejanza química con la sustancia mineral del hueso. En este trabajo se investigó la viabilidad de fabricación de andamios de CHAp/β-TCP mediante transformación pseudomórfica de bloques de yeso previamente moldeados. La transformación se realizó sumergiendo el bloque precursor de yeso en una disolución 1 M (NH4)2HPO4/1,33 M NH4OH en proporción líquido/sólido de 10 mL/g, y autoclavando a 120 °C y 203 kPa (2 atm) durante al menos 3 h. Ni la forma ni las dimensiones de la pieza cambiaron significativamente durante la transformación. La composición de los andamios obtenidos con 3 h de tratamiento fue 70% m/m CHAp y 30% m/m β-TCP, y sus resistencias a la compresión y a la compresión diametral fueron de 6,5 ± 0,7 y 5,3 ± 0,7 MPa, respectivamente. Incrementando el tiempo de tratamiento a 6 h, la composición del andamio se enriqueció en β-TCP (60% m/m CHAp y 40% m/m β-TCP), pero su resistencia no varió significativamente (resistencia a la compresión 6,7 ± 0,9 MPa y resistencia a la compresión diametral 5,4 ± 0,6 MPa). Sobre la base de los resultados obtenidos se concluyó que esta ruta resulta adecuada para la fabricación de andamios bifásicos a partir de piezas de yeso previamente moldeadas.
Modern approach to bone regeneration is based on the use of scaffolds and osteoinductive factors. Scaffolds are temporary tridimensional and macroporous structures made of biocompatible and resorbable materials which are intended to attract and support the colonization of mesenchymal stem cells and their differentiation into osteoproductive cells. Ideally, the scaffold resorption rate should match the rate of new bone growth ([1]).
The use of additive manufacturing (AM) processes for the fabrication of 3D scaffolds for bone regeneration has received great attention in the last decade ([2]). Among the several AM techniques available 3D-Printing (3-DP) on powder layer is particularly interesting because of its simplicity and low cost ([3]).
Some of the available commercial systems employ a powder mainly consisting in Plaster of Paris (CaSO4·(1/2)H2O, CSH) and an aqueous binder ink. During printing, the setting of powder takes place according to the hydration reaction of Eq. (1) and, after removing unreacted powder; solid pieces made of gypsum (CaSO4·2H2O, CSD) are obtained.
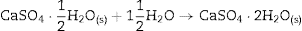
The in vivo resorption rate of CSD is considerably higher than the rate of growth of new bone. This is the main concern for using CSD scaffolds for bone regeneration procedures in clinics, especially when large defects are involved. Instead, materials with lower resorption rates, such as the less soluble apatite-like calcium phosphates, are generally preferred for manufacturing scaffolds for bone engineering ([7], [8], [9], [10], [11], [12], [13], [14]).
Pseudomorphing is the phenomenon by which one mineral is replaced by another while preserving the macroscopic shape, but changing the chemical composition and microstructure. It occurs in nature as consequence of weathering during millions of years ([15]). At the beginning of the seventies, coral (CaCO3, calcite) was pseudomorphically transformed into HAp by hydrothermal reaction with aqueous (NH4)2HPO4 at 180–350 °C and 103 MPa for 12–48 h ([16]). Since then, a lot of coralline HAp-based materials became commercially available and has been successfully applied for bone regeneration procedures in clinics.
More recently, the pseudomorphical transformation of CSD monoliths into HAp by treatment in alkaline phosphate solutions have been reported ([17], [18], [19], [20], [21], [22]). According to the published results, previous dehydration of CSD to CSH is required in order to strengthen the scaffold and to speed off the hydrothermal reaction. Minor amounts of more acidic calcium phosphates (CaHPO4·2H2O, DCPD or CaHPO4, DCP) were formed along with HAp.
The purpose of this study was to investigate the manufacturing of calcium phosphate scaffolds by direct hydrothermal transformation of CSD monoliths, without previous dehydration to CSH, and avoiding formation of acidic calcium phosphates.
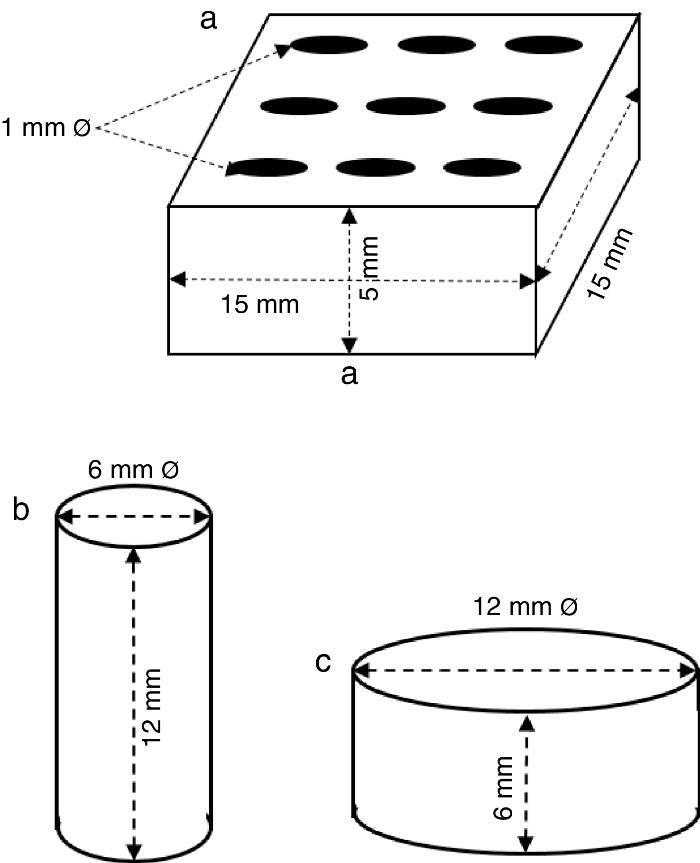
ExperimentalPreparation of CSD scaffolds and probesCSH (Gesso Diamante – Orto, IGE, Araripina, PE, Brazil) was mixed with water at W/P ratio of 0.50 mL/g to prepare scaffolds and probes with the shapes and sizes of Fig. 1.
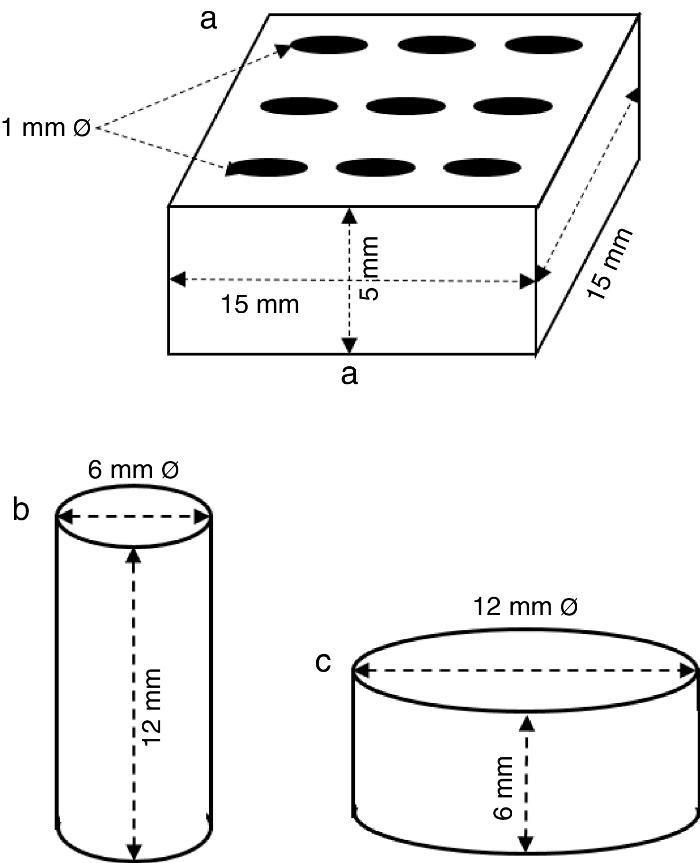
Fig. 1. Shapes and sizes of scaffolds and probes used in this work.
The holes in prismatic scaffolds (Fig. 1a) were made during molding. For this a silicone mold with inserts for 1 mm Ø stainless steel pins was employed. The slurry made from CSH and water (W/P = 0.5 mL/g) was casted in the assembled mold with inserted pins. When the initial setting was reached, pins were carefully removed and the piece was let setting for 1 h. Then, pieces were demolded and let dry in air to constant weight, and used for X-ray diffraction, SEM and EDS analysis.
Solid cylinders (Fig. 1b) and disks (Fig. 1c) (with not holes) were casted in a similar way, and used for strength and bioactivity tests.
Hydrothermal treatment with ammonium phosphate solutionGroups of 5 pieces were placed into glass flasks with (NH4)2HPO4/NH4OH (Vetec, Sao Paulo, Brazil) solution (1.00 M/1.33 M, pH 9.8) with a L/S ratio of 10 mL/g. The flasks were partially stoppered and autoclaved (Autoclave Vertical, CS, Prismatec, SP, Brazil) at temperature of 120 °C and pressure of 2 atm (203 kPa) for periods of time between 1 and 6 h. Then, autoclaved probes were washed with water and oven dried at 75 °C for 24 h.
Examination by XRD analysisX-ray diffraction (Shimadzu 7000, Japan) was used to analyze the qualitative and quantitative phase composition of the specimens before and after hydrothermal treatment. The X-ray diffraction patterns were obtained on powdered samples using CuKα (λ = 1.5418 ¿) radiation, and anode operation voltage and current of 40 kV and 30 mA, respectively. The 2θ interval between 10° and 40° was recorded at sweep rate of 0.2°/min. X’Pert HighScore Plus (PANalytical, The Netherlands) software was used for qualitative phase analysis and GSAS-II software ([23]) was employed to perform the quantitative phase analysis by the Rietveld method ([24]). The crystal structures of the ICSD database with codes 27875 for CSD, 73263 for CSH, 26205 for HAp and 97500 for β-tricalcium phosphate (β-TCP, β-Ca3(PO4)2) were used in Rietveld refining. The typical values obtained for the weighted profile R-factor during structural and compositional refining were in the range 11.3–12.2.
FTIR spectroscopyAttenuated Total Reflectance infrared spectra of powdered samples were collected in a Spectrum 400 FTIR/FTNIR spectrometer (Perkin Elmer, USA). Spectra were scanned in the range from 4000 to 650 cm−1 with resolution of 4 cm−1.
SEM observationExternal and fracture surfaces of untreated and treated with ammonium phosphate solution specimens were observed by a Phenom ProX (Phenom-World B.V., The Netherlands) desktop scanning electron microscope (SEM) coupled with an X-Ray Energy Dispersive Spectroscopy (EDS) system. Samples were examined as obtained, without any metal coating.
Measurement of compressive and diametral compressive strengthCompressive and diametral compressive strength tests were carried out on cylinders (12 mm h; 6 mm Ø; n = 5) and disks (6 mm h; 12 mm Ø; n = 5) Fig. 1), respectively, before and after being treated for 6 h. Tests were carried out in an Instron 3366 Dual Column testing machine (Instron Corp., USA) provided with 500 N and 10 kN load cells, for diametral and axial tests, respectively, and a plate displacement rate of 0.5 mm/min.
In vitro bioactivityThe in vitro bioactivity of selected samples, understood as the ability to form an apatite layer on their surfaces in vitro, was assessed as indicated in ISO 23317:2012 – Implants for surgery – In vitro evaluation for apatite-forming ability of implant materials ([25]). The test was conducted on disks (6 mm h; 12 mm Ø) by incubating in SBF at 36.5 ± 1.5 °C for 1, 3 and 7 days. The ratio of apparent surface of solid to SBF volume was 10 cm2/mL and flasks with samples soaked in SBF remained static during the whole incubation period. The presence of the apatite layer was verified by SEM.
Results and discussionThe scaffolds employed in this work (Fig. 1a) bore nine crossing holes distributed on their largest face to provide macroporosity, and to minimize the diffusion path of the reagent solution into the material.
The casted scaffolds were stiff enough to stand handling and water immersion without distinguishable disintegration.
The appearance of the scaffolds, before (as casted) and after hydrothermal treatment, is displayed in Fig. 2. Neither shape nor dimensions significantly changed after hydrothermal treating during different periods of time (1–6 h).

Fig. 2. Scaffolds before (as casted) and after hydrothermal treatment for 6 h in diammonium hydrogen phosphate solution.
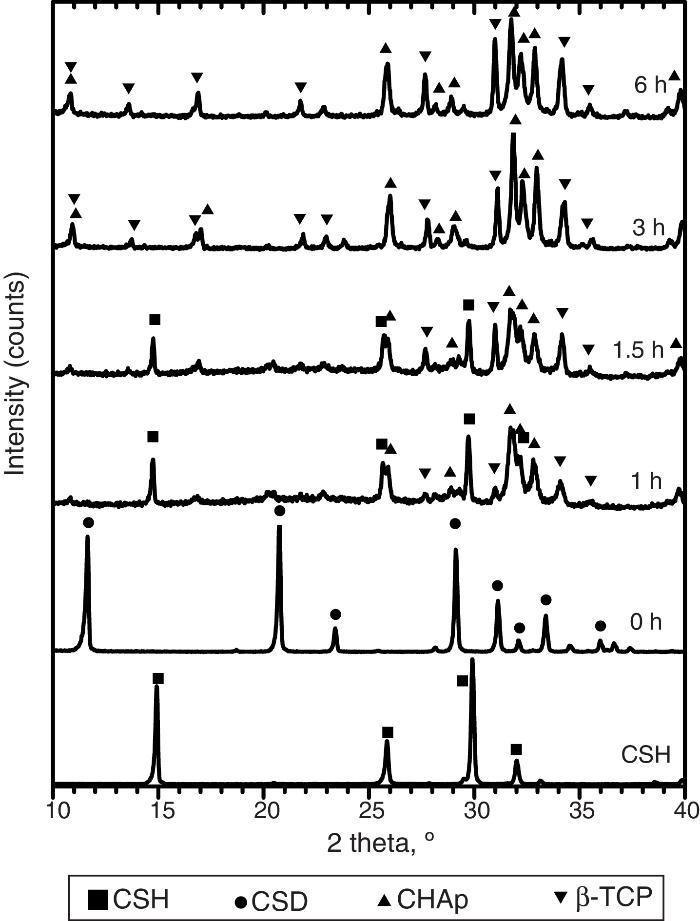
Only CSH peaks were observed in the X-ray diffraction pattern of the starting Plaster of Paris. On the other hand, CSD was the only crystalline phase found in casted scaffolds (Fig. 3, 0 h).
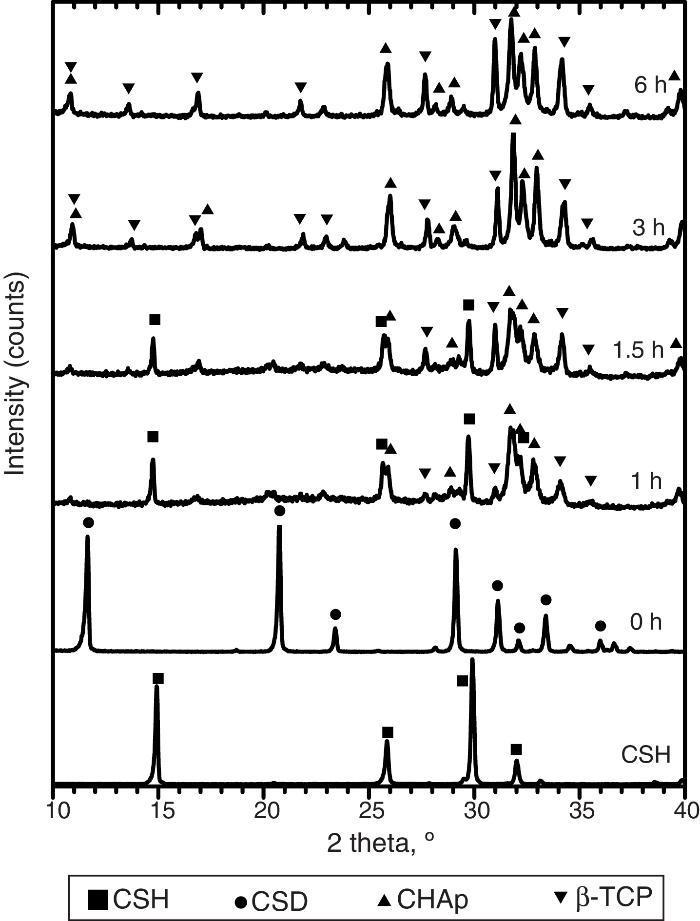
Fig. 3. Experimental X-ray diffraction patterns of the CSD scaffolds after different times of hydrothermal treatment.
During hydrothermal treating several changes in phase composition occurred which are displayed in the X-ray diffraction patterns of Fig. 3.
After 1 h of treatment only diffraction peaks corresponding to CSD, HAp and β-TCP were observed (Fig. 3, 1 h). Dehydration of CSD to CSH, as represented in Eq. (2), is to be expected since the CSD↔CSH transition temperature at 1 atm, is 97 °C ([26]), and the reaction mixture was submitted to temperature of 120 °C and vapor pressure of 2 atm.
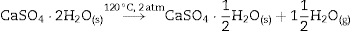
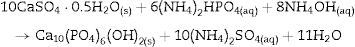
The formation of DCP and HAp in the core and the crust of CSD monoliths, respectively, during hydrothermal treatment at 120 °C in unbuffered (NH4)2HPO4 solution, has been previously described ([28]), and we can hypothesize that in the buffered solution employed in this work, DCP is formed in the core as an intermediate that continues reacting to β-TCP as exemplified in Eqs. eq0020, eq0025.
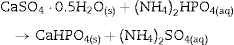
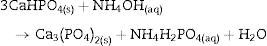
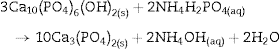
The same phases were observed for the period of 1.5 h (Fig. 3, 1.5 h).
After 3 h of treatment hydrothermal peaks corresponding to CSH completely disappeared and only HAp and β-TCP were detected (Fig. 3, 3 h). The estimated composition was 73 wt.% HAp and 27 wt.% β-TCP.
The picture change little at the end of 6 h of treatment (Fig. 3, 6 h) when 63 wt.% HAp and 37 wt.% β-TCP were obtained.
The reader should notice that in most of the previous reports the formula Ca10(PO4)5(OH)2 have employed to represent the apatite resulting from the hydrothermal reaction of Eq. (3). However, Suzuki et al. found that carbonate apatite was obtained by hydrothermal treatment of gypsum with (NH4)2HPO4 solution, even without intentional addition of any source of CO32− ion ([18]). They concluded that the alkaline character of the reaction media was responsible for absorption of CO2 from air which was incorporated to the structure of the apatite partially substituting PO43–, resulting in type B carbonate HAp (CHAp) ([18]).
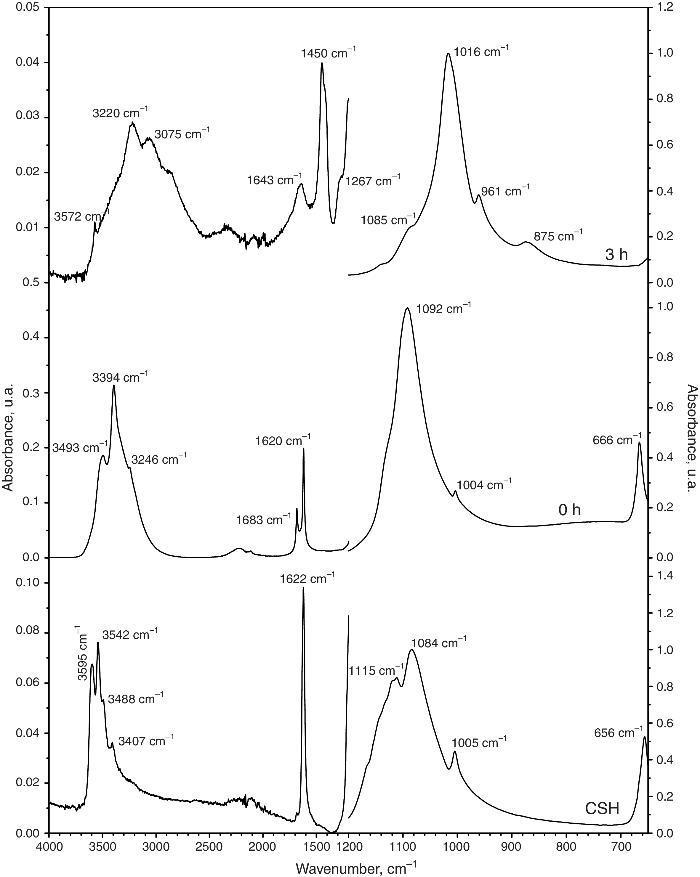
Fig. 4 shows the ATR-FTIR spectra of the starting powder of CSH, the CSD scaffolds immediately after setting (0 h), and after hydrothermal treatment for 3 h.
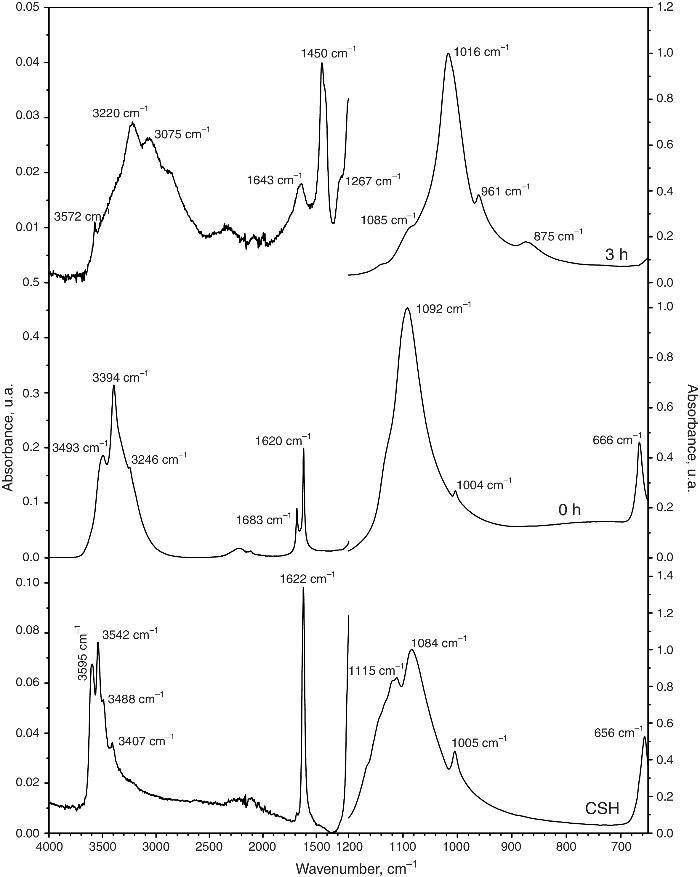
Fig. 4. ATR-FTIR spectra of the Plaster of Paris powder employed in the preparation of the scaffolds (CSH), gypsum scaffolds before hydrothermal treatment (0 h) and scaffolds hydrothermally treated for (3 h).
Bands corresponding to vibration stretching of the bond O–H of crystal water in the wavenumber region of 3600–3200 cm−1, and 3600–3000 cm−1 for the starting CSH powder and set scaffolds before any treatment (0 h). The single band at 1622 cm−1 and the doublet at 1683 and 1622 cm−1 are characteristic of bending vibration of crystal water in CSH and CSD, respectively. Typical strong and broad bands of stretching vibration of the S–O bonds in SO42− ion were observed in the region between 1200 and 950 cm−1 while bending vibrations appeared at 656 cm−1 and 666 cm−1 for CSH and CSD (0 h) ([30]). After the hydrothermal treatment for 3 h the bands characteristic for vibrations of SO42− group and crystal H2O in CSD were replaced by vibrational bands corresponding to vibrations characteristic of free OH, absorbed H2O, PO43− and CO32−. The acute band at 3572 cm−1 is characteristic of the stretching vibration of the structural OH group in HAp and the other bands in the region of 3200–3000 cm−1 and the band at 1643 cm−1 can be assigned to the stretching and bending vibrations of OH in loosely bonded water, respectively. Asymmetric (ν3) and symmetric (ν1) stretching vibrations of P-O bond in PO43− are the responsible for the bands at 1000–1100 cm−1 and 961 cm−1, respectively ([31], [32]). The bands characteristic of asymmetric stretching vibration (ν3) and out-of-plane bending of the C-O bond in CO32− group in carbonate HAp (CHAp) are observed at 1448 cm−1 and 875 cm−1, respectively ([33]). The ν3 band at 1450 cm−1 can considered as a fingerprint for type B CHAp ([34]), that is, part of the PO43− ions in the crystal structure was replaced for CO32− ions.
Thus, the product of the hydrothermal treatment of CSD scaffolds was a mixture of type B CHAp and β-TCP.
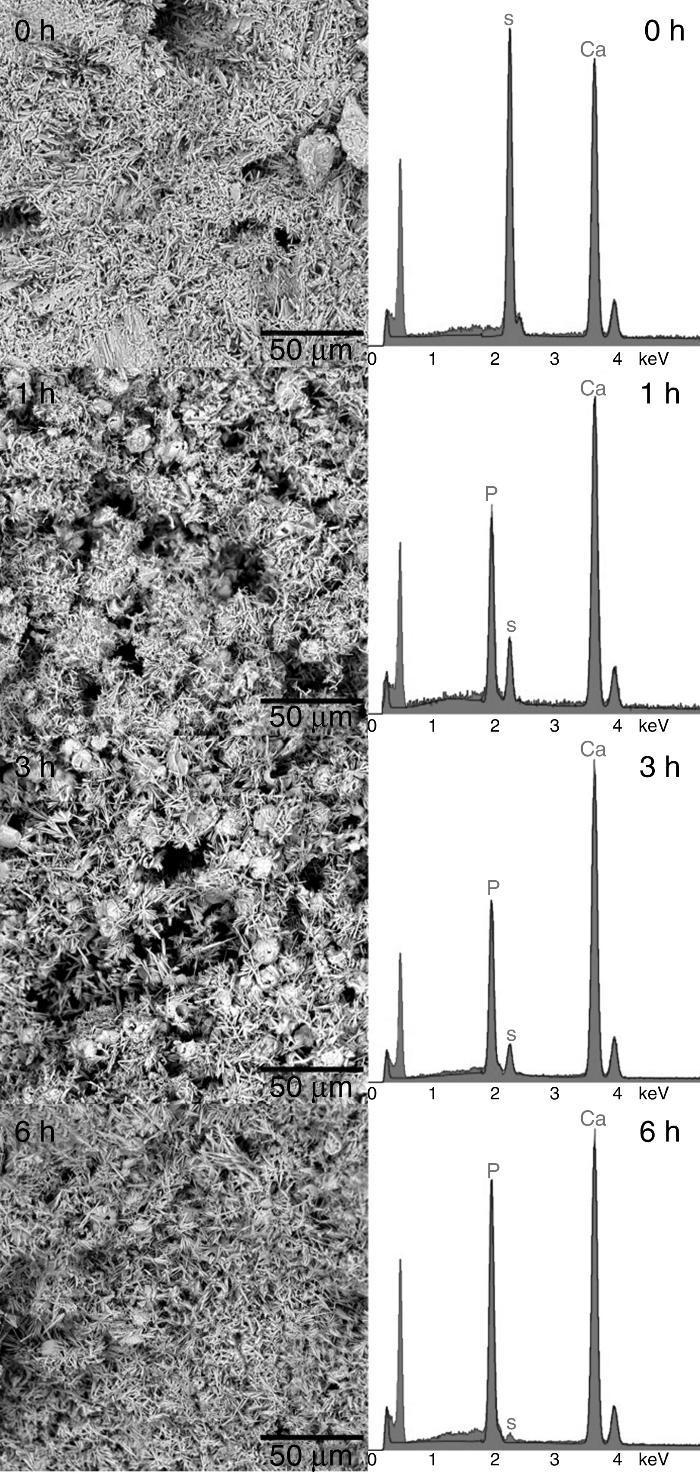
Structural and compositional modifications experienced by the CSD scaffolds after 1, 3 and 6 h of treatment are displayed in Fig. 5. The fracture surface of CSD scaffolds before treatment consisted of needle- and plate-like crystals typical of CSD ([18]), being more abundant the first ones. The EDS analysis confirmed that only Ca, S, and O occurred (Fig. 4, 1 h) as corresponds for CSD.
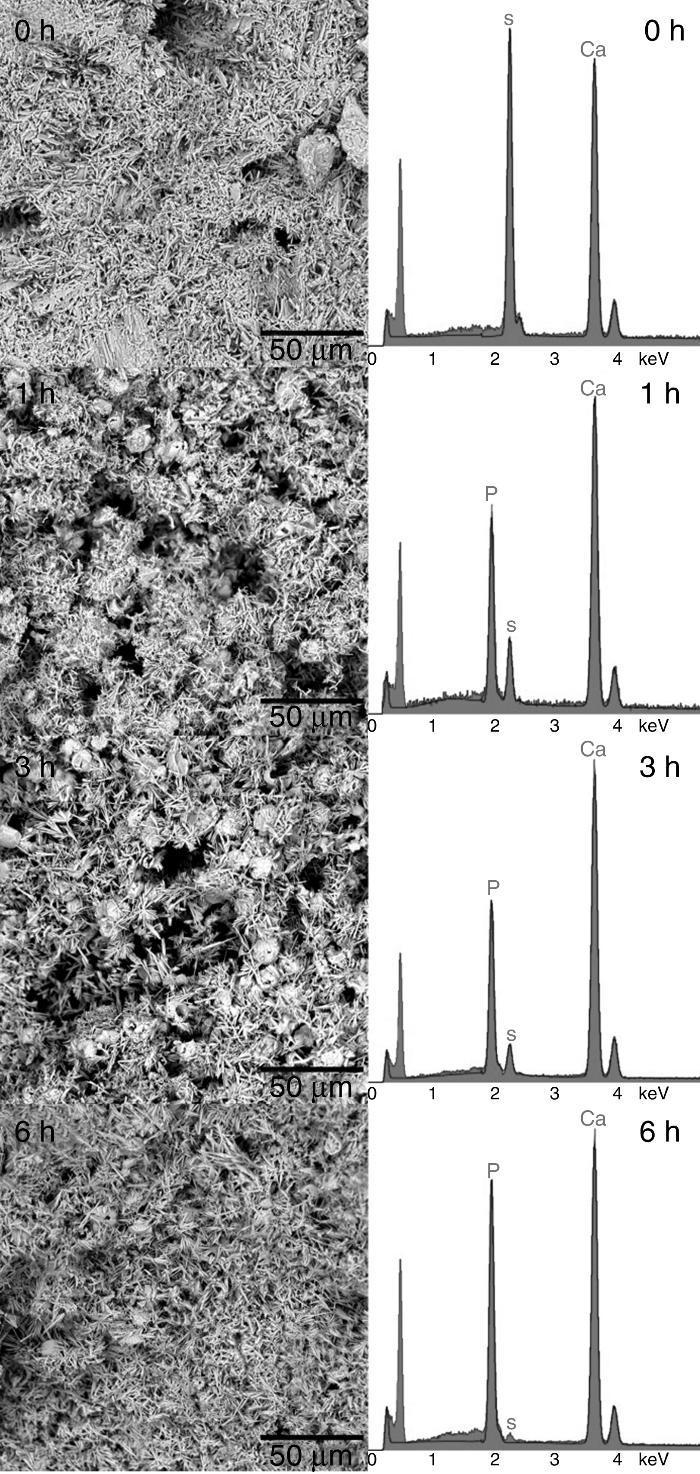
Fig. 5. SEM micrographs of fracture surface of the CSD scaffolds after treatment for 0, 1, 3 and 6 h in 1 M (NH4)2HPO4/1.33 M NH4OH.
In the SEM micrograph of the specimen treated for 1 h an increase in porosity could be noticed with regard to the untreated scaffold. Its structure consisted of fine needle-like crystals of HAp ([18]) together with prismatic crystals typical of β-TCP ([35]). The appearance of Ca and P peaks, and the diminution of S peak in the EDS spectrum confirmed the formation of Ca,P-containing phases at the expense of CSH (Fig. 4, 1 h).
By increasing the transformation time to 3 and 6 h, little change was observed in morphology of the scaffolds. However, the EDS spectra showed continued decreasing of the intensity of the S peak and increasing of Ca and P peaks. A decrease of the intensity ratio of Ca and P peaks could be noticed as well, from 3 to 6 h, caused by the decrease in HAp/β-TCP abundance ratio, as already evidenced from the results of quantitative phase analysis.
The above results showed that biphasic calcium phosphate scaffolds consisting of CHAp and β-TCP, were obtained after 3 h of treatment by the procedure described above.
The CHAp:β-TCP ratio can be increased by using longer periods of treatment. For example, hydrothermal treatment for 3 h yielded scaffolds with CHAp:β-TCP ratio of 70:30, approximately; while after 6 h of treatment CHAp:β-TCP ratio was 60:40. Controlling the CO3-HAp:β-TCP ratio allows tuning the resorption rate of the scaffolds, in similar way as in commercial biphasic calcium phosphate ceramics (BCP) constituted of HAp and β-TCP ([36]).
Probes of CSD treated as described before for 3 h exhibited compressive strength (CS) of 6.5 ± 0.7 MPa. During compressive testing, upon reaching the limit load, the test probes were crushed instead of breaking fragilely. In order to indirectly assess the tensile strength, diametral compressive (DCS) test was also carried out. In this test a tensile stress is generated normally to the loading direction on a vertical disk ([37]). A DCS of 5.3 ± 0.7 MPa was found for probes submitted to hydrothermal treatment for 3 h. This value was significantly greater than the value of 2.8 MPa reported for CSD monoliths treated with (NH4)3PO4 at 100 °C for 24 h ([19]).
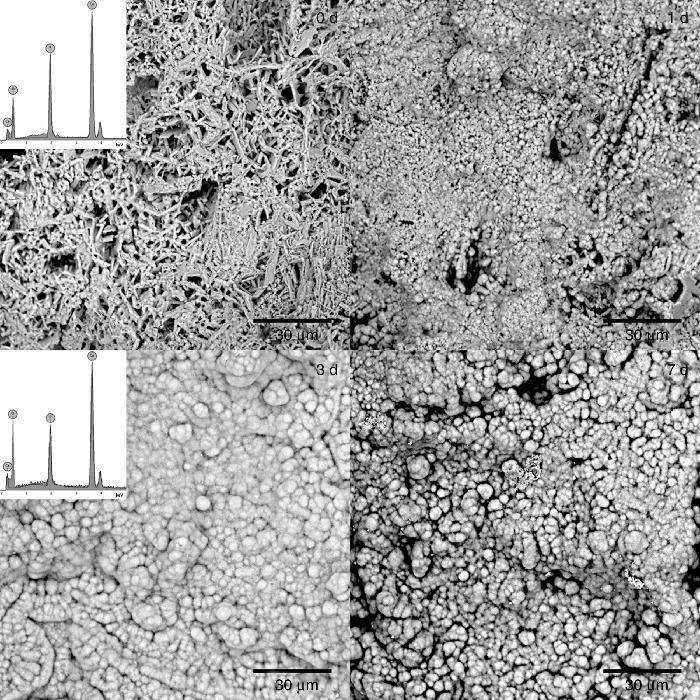
The formation of a surface layer of biological carbonate-apatite when implanted in vivo, is a necessary condition for bonding of bioactive materials to living bone tissue. In vivo bioactive materials are also able to form a surface layer of apatite after being immersed in a synthetic solution that imitates the composition of blood plasma (SBF), at body temperature, for different periods of time, and these are the basis for a common in vitro method to determine bioactivity of materials ([25], [38]). Thus, in vivo bioactivity was determined for the CHAp/β-TCP scaffold obtained by 3 h of treatment. The SEM micrographs of the sample surface after soaking for 0, 1, 3 and 7 days are shown in Fig. 6. After 1 day of immersion in SBF it was detected a change in the surface morphology of the sample, characterized by the appearance of globular crystalline aggregates. The globular aggregates grew and reached confluence after 3 days of immersion, completely covering the surface of the sample, and this morphology was maintained after 7 days of immersion. The same structure has been described for the apatite-layer formed by bioactive materials when soaked in SBF ([38]). The EDS analysis of the surface of samples revealed the presence of Ca, P and O, before soaking (0 d) and after 3 days of soaking in SBF (3 d), but the Ca/P peak intensities increased with soaking. Bearing in mind that before soaking there is a mixture of approximately 70 wt.% CHAp and 30 wt.% β-TCP, the increasing of the Ca/P ratio by soaking points out that globular aggregates consist of apatite, and that the scaffold obtained is bioactive in vitro.
Fig. 6. SEM micrographs of the surface of scaffold prepared by 3 h of treatment and immersed in SBF at 36.5 °C for 0, 1, 3, and 7 days. Inserts: EDS spectra of scaffolds soaked 0 and 3 days in SBF.
According to authors’ best knowledge, obtaining a mixture of CHAp and β-TCP by hydrothermal treatment of gypsum monoliths has not been previously described. While the formation of HAp, DCP or DCPD, and mixtures of HAp and DCP (or DCPD) has been previously reported ([22], [28], [39]), the formation of β-TCP by hydrothermal reaction of CSD in solutions of ammonium phosphate was no observed. All researchers who have addressed the transformation of CSD into calcium phosphates have employed plain solutions of (NH4)2HPO4, regardless of the acidification of the reaction medium that occurs, as illustrated in Eqs. eq0015, eq0020, eq0025, during the transformation of gypsum. This can result in the formation of acidic phases such as DCP or DCPD. The consequences of the acidification of the medium can be most evident in the core of the monolith, where removal of exhausted medium and arrival of fresh one can be seriously hindered ([28], [40]). The reaction medium employed in this work was a solution containing 1 M (NH4)2HPO4 and 1.33 NH4OH with high buffer capacity which ensures the maintenance of a pH higher than 9 along the complete reaction. However, the mechanism conducting to the formation of β-TCP deserves further research.
ConclusionsScaffolds containing two crystalline phases, CHAp and β-TCP, where obtained by the method proposed in this work, using previously casted CSD monoliths as starting material. Complete transformation of the precursor was achieved after 3 h of reaction. The ratio between the constituent phases (CHAp and β-TCP) can be varied adjusting the reaction time: The larger the reaction time, the richest is the scaffold in β-TCP. Neither shape nor dimensions were significantly affected during processing, and the resulting biphasic scaffolds were strong enough to stand handling and immersion in water. The scaffolds developed a surface apatite layer after soaking 3 days in SBF which suggest they are bioactive in vivo.
The processing route proposed is effective for manufacturing biphasic (CHAp/β-TCP) scaffolds from previously shaped pieces of gypsum.
Acknowledgements
The authors acknowledge to CAPES, Brazil for supporting H.A. Batista MSc studies, to CNPq, Brazil for financing DCR Process No. 351182/2013-6, and PVE Process 314532/2014-5, and to the Ministry of Economy and Competitiveness, Spain for financing project BIOJER (MAT2013-48426-C2-1-R).
Received 14 February 2016
Accepted 22 February 2016
Corresponding author. husonab@live.com

