




Article information
Full Text
Bibliography
Download PDF
Statistics
Tables (2)

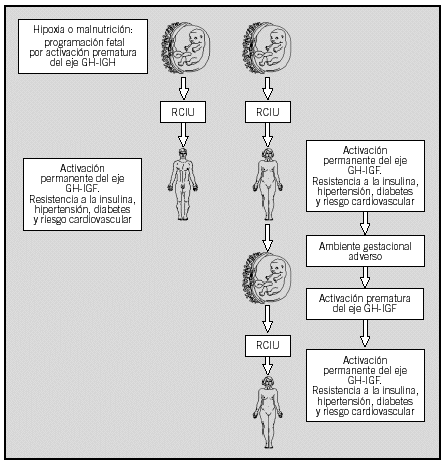
Fig. 1. La agresión inicial genera la activación del eje GH-IGF, que desencadena la programación fetal que tendrá consecuencias en la vida adulta. En la mujer afectada crearán un ambiente adverso al feto en desarrollo que pondrá nuevamente en activación el eje y perpetuará la programación. En el varón sólo se presentarán las consecuencias de la programación.GH-IGF: hormona de crecimiento; factor de crecimiento insulinoide.RCIV: retraso del crecimiento intrauterino.
Show moreShow less
Article
These are the options to access the full texts of the publication Medicina Clínica
Subscriber
Subscribe

Purchase
Contact
Phone for subscriptions and reporting of errors
From Monday to Friday from 9 a.m. to 6 p.m. (GMT + 1) except for the months of July and August which will be from 9 a.m. to 3 p.m.
Calls from Spain
932 415 960
Calls from outside Spain
+34 932 415 960
E-mail
