




Familial non-medullary thyroid cancer is defined as the presence of non-medullary thyroid cancer in two or more first-degree relatives, in the absence of other predisposing factors. It represents up to 9% of differentiated thyroid cancers, and only a minority appears in well-known hereditary syndromes that associate thyroid cancer among many other clinical manifestations. However, in more than 95% of cases, thyroid cancer appears isolated, and its genetic causes have yet to be elucidated. We review here the current knowledge of the genetic basis of this pathology, as well as its clinical characteristics. Understanding the genetic mechanisms implied would help to comprehend the metabolic pathways involved, with the consequent potential therapeutic application. In addition, it would allow genetic counseling and to focus our efforts on patients at risk of developing this disorder.
El carcinoma diferenciado de tiroides familiar se define como la presencia de cáncer de tiroides no medular en dos o más familiares de primer grado, en ausencia de otros factores predisponentes. Representa hasta el 9% de los cánceres diferenciados de tiroides, y solo una minoría aparece en el seno de síndromes hereditarios bien conocidos que asocian el cáncer de tiroides entre otras múltiples manifestaciones clínicas. Sin embargo, en más del 95% de los casos, el cáncer de tiroides aparece de manera aislada, y sus causas genéticas aún están por dilucidar. En esta revisión, resumimos el estado actual del conocimiento de las bases genéticas de esta patología, así como sus características clínicas. La comprensión de los mecanismos genéticos implicados ayudaría a esclarecer las vías metabólicas involucradas, con la consiguiente potencial aplicación terapéutica. Además, permitiría ofrecer consejo genético y focalizar nuestros esfuerzos en los pacientes susceptibles de padecer la patología.
Thyroid cancer is the most common cancer of the endocrine system, and is currently the fifth most common cancer in women in the United States.1 Follicular cell-derived differentiated thyroid carcinomas (DTC) account for 95% of thyroid cancers. These include: papillary carcinomas (approximately 80% of cases); follicular carcinomas (15%); and other less common types (<5%). The remaining 5%, medullary thyroid carcinomas (MTC), originate from C or parafollicular cells, which produce calcitonin.2
In some cases, several members of the same family have thyroid cancer. An integrated care approach, which may include genetic counselling, requires an understanding of the genetic bases of the disease. In the case of familial MTC (up to 25% of MTC), it is known that the RET proto-oncogene is the main protagonist involved.3 The presence of germline RET mutations is associated with hereditary MTC or type 2 multiple endocrine neoplasia, with an autosomal dominant inheritance pattern (ADI). A great deal has been learned in recent years about the genetics of this disease, even establishing a close genotype-phenotype correlation.4 However, in this review our focus is on familial non-medullary thyroid carcinoma, whose hereditary basis is much less well understood.
The first step is to define the disease: familial differentiated (or non-medullary) thyroid carcinoma, also known as FNMTC (familial non-medullary thyroid cancer), is defined by the presence of this cancer in two or more first-degree relatives, provided there are no other predisposing factors such as radiation or iodine deficiency.5
The second is to be aware that the prevalence of FNMTC is not negligible: it is estimated that up to 9% of non-medullary thyroid carcinomas have a hereditary component.5 Therefore, it is sufficiently common that we are likely to come across some cases in our clinical setting. However, it is also sufficiently uncommon that we have to join forces with multiple other centres to gather a large enough sample of cases to further research the disease.
The third is that once a possible FNMTC has been identified, we then have to distinguish between syndromic and non-syndromic cases. In other words, whether the FNMTC has occurred in isolation or is part of the phenotypic spectrum of a syndrome. In 95% of cases, the patient will have a non-syndromic FNMTC. However, in around 5%, the FNMTC is a component of a well-characterised genetic syndrome.6 We have to be aware of this possibility due to the potential systemic repercussions, as a multidisciplinary approach will be necessary.
The aim of this review was to encapsulate what is known to date about the genetic bases of FNMTC, focusing on the non-syndromic type. We carried out a literature search in the MEDLINE database and selected the most relevant original reviews or articles published in the last 20 years (1999–2020) on this topic. The keywords used in the search were “thyroid cancer”, “familial”, “genetics”, “germline mutations” and “non-medullary”.
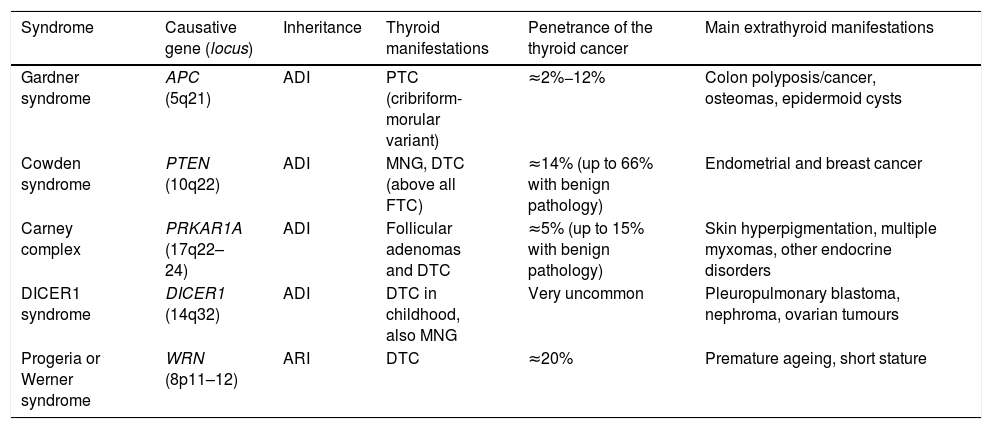
Syndromic familial non-medullary thyroid carcinomaThere are five syndromes currently recognised as occurring with FNMTC for which the genes involved have been identified. We provide a brief description of these syndromes below (Table 1). In other syndromes, such as McCune-Albright, Peutz-Jeghers and ataxia-telangiectasia, a predisposition to differentiated non-medullary thyroid cancer has also been suggested but has not been clearly established.
Hereditary syndromes associated with FNMTC.
| Syndrome | Causative gene (locus) | Inheritance | Thyroid manifestations | Penetrance of the thyroid cancer | Main extrathyroid manifestations |
|---|---|---|---|---|---|
| Gardner syndrome | APC (5q21) | ADI | PTC (cribriform-morular variant) | ≈2%−12% | Colon polyposis/cancer, osteomas, epidermoid cysts |
| Cowden syndrome | PTEN (10q22) | ADI | MNG, DTC (above all FTC) | ≈14% (up to 66% with benign pathology) | Endometrial and breast cancer |
| Carney complex | PRKAR1A (17q22–24) | ADI | Follicular adenomas and DTC | ≈5% (up to 15% with benign pathology) | Skin hyperpigmentation, multiple myxomas, other endocrine disorders |
| DICER1 syndrome | DICER1 (14q32) | ADI | DTC in childhood, also MNG | Very uncommon | Pleuropulmonary blastoma, nephroma, ovarian tumours |
| Progeria or Werner syndrome | WRN (8p11–12) | ARI | DTC | ≈20% | Premature ageing, short stature |
ADI: autosomal dominant inheritance; ARI: autosomal recessive inheritance; DTC: differentiated thyroid carcinoma; FTC: follicular thyroid carcinoma; MNG: multinodular goitre; PTC: papillary thyroid carcinoma.
Gardner syndrome is an ADI syndrome, caused by germline mutations in the APC tumour suppressor gene (chromosome 5q21). It is a variant of familial adenomatous polyposis, in which there are also extraintestinal manifestations. Clinical manifestations include colon polyposis/cancer, osteomas, epidermoid cysts and dermoid tumours. Some 2%−12% of patients can develop DTC, usually at a young age and, in up to a third of cases, as the first manifestation of the syndrome. It is associated with the cribriform-morular variant of papillary thyroid carcinoma, so for any patient with this histology we have to ensure we rule out Gardner syndrome.7
Cowden syndrome [MIM: 158350]This is an ADI syndrome associated with inactivating germline mutations in the PTEN suppressor gene (chromosome 10q22). Patients suffer from multiple hamartomas and an increased risk of endometrial and breast cancer, and as many as two out of every three patients have thyroid disease, mainly multinodular goitre (MNG) and DTC (the latter of the two in 3%–14% of cases, and mainly of follicular lineage).7 In patients in whom PTEN mutations are not found (up to 15%), other genes less commonly associated with Cowden syndrome should be considered, such as SDHB-D, KLLN promoter hypermethylation8; PIK3CA and AKT19; RASAL110 and SEC23B.11
Carney complex [MIM: 160980]This is an ADI syndrome due to germline mutations in the PRKAR1A suppressor gene (chromosome 17q22–24), which causes skin hyperpigmentation (lentigines and blue naevi in particular), myxomas in multiple locations, and endocrine, pituitary, adrenal, gonadal and/or thyroid disorders. Up to 15% of those affected may have adenomas and, more rarely, thyroid carcinomas. It should not be confused with the term “Carney's triad” (association of gastrointestinal stromal tumour [GIST] + pulmonary chondroma + paraganglioma).7
DICER1 syndrome [MIM: 606241]This is an ADI syndrome, described for the first time more recently, related to germline mutations in DICER1 (chromosome 14q32), which encodes a ribonuclease, predisposing to multiple childhood tumours such as pleuropulmonary blastoma, as well as multinodular goitre (MNG) and DTC. This gene should particularly be taken into account in paediatric thyroid cancers.12
Progeria or Werner syndrome [MIM: 277700]This syndrome, caused by mutations in the WRN gene (chromosome 8p11–12), is the only one of the autosomal recessive inheritance (ARI) syndromes to encode a helicase involved in telomere maintenance. Its clinical characteristics include short stature, multiple neoplasms (including thyroid, in up to 20% of cases), and the most distinctive: premature ageing. It is very rare outside Japan.7
Non-syndromic familial non-medullary thyroid carcinomaMost of our patients with thyroid cancer have non-medullary histological subtypes (mainly papillary or follicular carcinomas), with approximately 9% having a family history. Apart from the few that occur as components of the aforementioned syndromes, the majority are non-syndromic, and their genetics have yet to be determined. We discuss the clinical characteristics of non-syndromic FNMTC below, and then focus on the susceptibility genes described to date.
Clinical characteristicsFNMTC, which accounts for 3.2%–6.2% of all thyroid cancers, was first recognised as a disease in its own right over ten years ago.5 However, with the high prevalence and incidence of thyroid cancer in the general population (15.8 new cases/100,000 population/year are diagnosed13), we have to ask whether the presence of two or more cases of non-medullary thyroid cancer in the same family might not simply be down to chance. After a complex statistical analysis, Charkes14 concluded that around 62% of families with two cases may be phenocopies (two sporadic cases associated by chance). However, if there are three affected members, there is a 96% likelihood that it really is hereditary.
FNMTC has been found to have ADI, with variable expressivity and incomplete penetrance. The majority of FNMTC are papillary carcinomas (90%), followed by follicular carcinomas, as in sporadic non-medullary carcinomas. Different histological variants can coexist within the same family. In terms of somatic molecular alterations, when comparing thyroidectomy/fine needle aspiration biopsy (FNAB) samples from patients with FNMTC to those from DTC patients with no family history, no significant differences were found in terms of frequency or type of somatic mutations, with presence of the BRAF Val600Glu mutation, mutations in NRAS or RET/PTC rearrangements in very similar percentages.6 This supports the theory of germline mutations causing the disease, without somatic mutations playing a crucial role in tumourigenesis.
Overall, the clinical characteristics are similar, with a female predominance in some studies (2−3:1 ratio). As differential features, age at diagnosis tends to be younger in FNMTC, at around 43 years compared to 48 in sporadic DTC.6 Recent studies even suggest that the second generation of FNMTC sufferers may show a “phenomenon of anticipation”, with diagnosis at a younger age and more aggressive disease than in the previous generation, although we cannot rule out that it may be partly due to earlier detection in these people because of their family history. There would also be a higher proportion of affected males.15,16
A 2015 meta-analysis that included 12 studies with fairly disparate results also described the cancer as being more aggressive in FNMTC.17 The authors found more lymphadenopathy and extrathyroid spread, a higher rate of multifocality and recurrence, and shorter progression-free survival, especially in the studies which included families with at least three affected members; the differences in families with only two affected members were less clear. Along the same lines, Ya-Bing Zhang et al.18 described greater aggressiveness in families with three or more members with FNMTC, suggesting that studies should perhaps focus on these families with a larger number of affected patients.
The utility of carrying out DTC screening has been investigated in people with relatives who suffer from the disease.19 In this prospective five-year study, DTC was detected by screening in 4.6% of at-risk individuals from families with two affected members, and in 22.7% from families with three or more affected members, leading to the conclusion that active surveillance should be considered in the second of the two groups. In other words, the more people affected, the greater the risk of more family members having the disease, and it would be more appropriate to carry out active surveillance.20 It has also been documented that the FNMTC detected by screening had a smaller tumour size, a lower rate of lymphadenopathy and resulted in less aggressive surgical and radioactive iodine therapy than the index cases.19 Nevertheless, the latest DTC management guidelines of the American Thyroid Association (ATA)21 do not take a position for or against family screening, as no reduction in morbidity or mortality has been demonstrated. In reality, no such reduction is likely to be demonstrated in a disease with a 10-year survival rate of 95%–97%. Triponez et al.22 compared the survival of patients with FNMTC with that of their unaffected relatives and with the general population, and found shorter survival in patients whose families had three or more affected members. However, they did not compare their figures with sporadic DTC.
In any case, most of the publications acknowledge that the greater aggressiveness of FNMTC is still a matter of debate due to the heterogeneity of the studies, which are mainly retrospective. The same lack of consensus is extrapolated to the follow-up and treatment of these tumours. It has recently been suggested that the initial surgical treatment of FNMTC should be more aggressive in families with three or more affected members.23 In fact, the latest DTC management guidelines21 advocate total thyroidectomy and radioactive iodine therapy in patients with a family history of DTC. One study also suggests that, as these patients may have more lymphadenopathy, a central lymphadenectomy should be considered in conjunction with total thyroidectomy.24 What would not be indicated is prophylactic total thyroidectomy, as we have not yet identified the genetic causes of the disease to enable carriers to be detected, and there is also incomplete penetrance. FNAB is currently recommended in nodular lesions over 1 cm in size25 (in the previous 2009 ATA guidelines, it was also recommended in nodules measuring less than a centimetre). The possibility of more false negative FNAB results in patients with FNMTC has been reported, as these patients tend to have a high incidence of multifocal malignant disease, coexisting with benign nodules.26
In summary, it seems reasonable to consider the presence of a family component as one more risk factor to take into account in the personalised management of thyroid cancer,27 particularly in families with three or more affected members. This is because of the high likelihood of it being hereditary and not by chance, but also in view of the studies showing greater tumour aggressiveness and greater benefit from early screening in this subgroup.
Genetic basesAlthough FNMTC is relatively rare, it represents a significant burden in terms of resources for the Spanish national health service, primarily because of the lack of knowledge about both the hereditary factors and the possible clinical differences compared to sporadic disease. These circumstances complicate early diagnosis (in stages where treatment is more decisive), and make it unfeasible to carry out genetic counselling which, in addition to the impact on healthcare, is increasingly in demand in today's society. Knowing the mutation involved in a genetic cancer would have the direct effect of enabling a family study, identifying members who are carriers of the mutation, concentrating diagnostic and therapeutic efforts on those people, and being able to influence the course of these cancers at an early stage. It would also avoid having to follow up all non-carrier family members clinically. Moreover, recent scientific history tells us that the identification of genes that cause hereditary cancers has led to huge advances in the understanding of oncogenesis in sporadic cases (much more prevalent), in the identification of components and cell signalling pathways involved, and in the design of molecules capable of modulating these pathways and exerting specific therapeutic actions. Therefore, knowledge of the genetic bases of a disease like FNMTC is important for the direct implications it could have on healthcare practice.
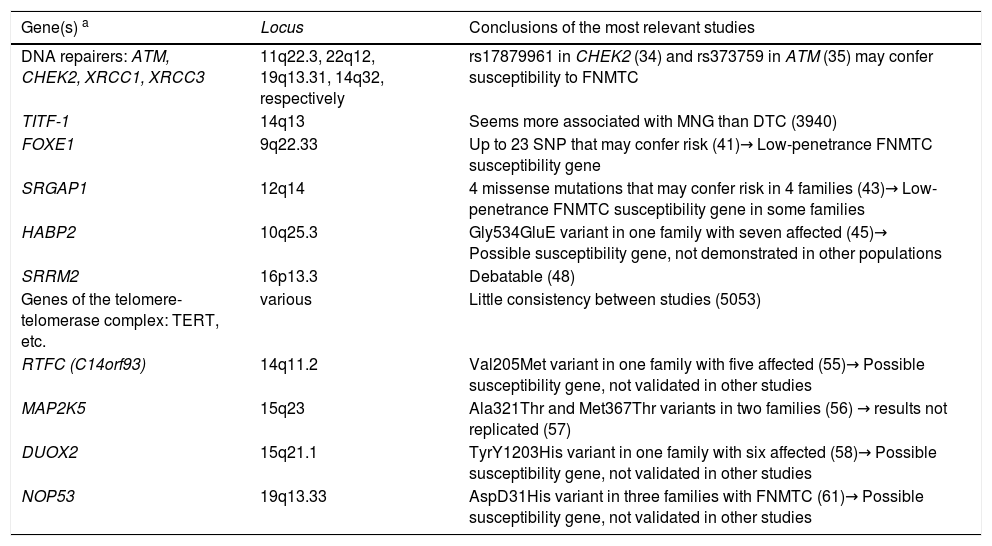
The different search strategies used for the genes involved in this disease are detailed below. Table 2 provides a summary of the main candidate genes currently identified, which are described later in this article.
Genes/families of genes described in non-syndromic FNMTC.
| Gene(s) a | Locus | Conclusions of the most relevant studies |
|---|---|---|
| DNA repairers: ATM, CHEK2, XRCC1, XRCC3 | 11q22.3, 22q12, 19q13.31, 14q32, respectively | rs17879961 in CHEK2 (34) and rs373759 in ATM (35) may confer susceptibility to FNMTC |
| TITF-1 | 14q13 | Seems more associated with MNG than DTC (3940) |
| FOXE1 | 9q22.33 | Up to 23 SNP that may confer risk (41)→ Low-penetrance FNMTC susceptibility gene |
| SRGAP1 | 12q14 | 4 missense mutations that may confer risk in 4 families (43)→ Low-penetrance FNMTC susceptibility gene in some families |
| HABP2 | 10q25.3 | Gly534GluE variant in one family with seven affected (45)→ Possible susceptibility gene, not demonstrated in other populations |
| SRRM2 | 16p13.3 | Debatable (48) |
| Genes of the telomere-telomerase complex: TERT, etc. | various | Little consistency between studies (5053) |
| RTFC (C14orf93) | 14q11.2 | Val205Met variant in one family with five affected (55)→ Possible susceptibility gene, not validated in other studies |
| MAP2K5 | 15q23 | Ala321Thr and Met367Thr variants in two families (56) → results not replicated (57) |
| DUOX2 | 15q21.1 | TyrY1203His variant in one family with six affected (58)→ Possible susceptibility gene, not validated in other studies |
| NOP53 | 19q13.33 | AspD31His variant in three families with FNMTC (61)→ Possible susceptibility gene, not validated in other studies |
Various strategies have been developed to search for candidate genes responsible for FNMTC. Prior to the emergence of modern sequencing techniques, the following had been carried out:
- a)
Linkage analysis. In genetics, linkage refers to the physical association between two loci (i.e. their closeness in the same DNA strand, which results in a low rate of recombination between them during meiosis and so a greater likelihood of joint inheritance). In related individuals, genetic loci are mapped to find inherited alleles linked to the disease of interest and to indirectly determine the chromosomal location of the gene associated with FNMTC. In other words, these types of study are an indirect method that uses genetic markers (microsatellites for example) linked to the disease gene.
The loci described to date include TCO28 (19q13.2), fPTC/PRN (1q21), FTEN (8p23.1-p22), NMTC1 (2q21), MNG1 (14q32), 6q22, 8q24 and 4q32.29 However, the candidate genes located at these loci remain unknown.
- b)
Genome-wide association (GWA) studies. Thousands of polymorphisms are studied, comparing controls to patients, in order to find predisposition polymorphisms. For example, two single nucleotide polymorphisms (SNP) associated with risk of FNMTC were identified using this technique: rs944289 (located at 14q13.3, near NKX2-1) and rs965513 (located at 9q22.33, close to the candidate gene FOXE1).30 However, most of the studies are carried out in non-familial DTC.31
- c)
Studies targeting candidate genes or specific metabolic pathways. For example, Pereira et al.32 directly studied FOXE1 in 60 families with FNMTC in Portugal, finding an association between germline variants of this gene and the development of FNMTC in these families.
However, the results from these studies did not prove particularly useful, describing genes that could be involved in the aetiology of FNMTC with low penetrance, and only in some families. Furthermore, the results have not been replicated in subsequent studies.
- d)
Next-generation sequencing techniques. In recent years, there have been great advances in the field of genomics thanks to the greater technical and economic feasibility of carrying out next-generation sequencing. Next-generation sequencing (NGS) brought about a huge leap from examining specific genes to exploring entire genomes. All this could also be done much more efficiently and rapidly than with the classic Sanger sequencing. It is now possible to compare, for example, how the genome of patients with a certain disease differs from the genome of healthy individuals. Since the incorporation of the new sequencing techniques, studies trying to determine the genetic causes of FNMTC have multiplied, identifying new genes which are compiled in a recent review.33 In the next section we review what is known about the genes described so far associated with vulnerability to FNMTC.
Genes involved in DNA repair: ATM (ATM serine/threonine kinase, locus 11q22.3), CHEK2 (checkpoint kinase 2, locus 22q12), XRCC1 (X-Ray repair cross complementing 1, locus 19q13.31) and XRCC3 (X-Ray repair cross complementing 3, locus 14q32).
The fact that factors such as ionising radiation increase the risk of thyroid cancer suggests that DNA repair genes could be involved in the pathophysiology. Genes such as ATM, CHEK2, XRCC1 and XRCC3 have therefore been studied. Several studies have reported that certain polymorphisms in these genes could be associated with greater susceptibility to DTC, such as rs17879961 in CHEK2,34 and rs373759 in ATM.35 Also, mutations in CHEK2 could be more common in cases of DTC.36 In contrast, Ryu et al.37 reported that the Arg194Trp change in XRCC1 conferred less susceptibility to DTC in the Korean population. These studies were essentially carried out comparing patients with sporadic DTC to controls,38 and no clear relationship with FNMTC can be established for the moment.
TITF-1 (thyroid transcription factor 1, also known as NKX2.1)A thyroid transcription factor is located at 14q13. In 2009, the Ala339Val germline mutation in TITF-1 was described in four patients with a history of MNG and papillary thyroid carcinoma, two of whom had a family history of MNG, with or without papillary thyroid cancer.39 A year later, Cantara et al.40 did not find the presence of this variant in TITF-1 in their series of patients with papillary thyroid carcinomas (PTC). Therefore, although the possibility of this variant being associated with MNG cannot be ruled out, it cannot be confirmed that it is related to PTC and even less so to FNMTC.
FOXE1 (forkhead box E1)It is located at 9q22.33 and encodes a thyroid transcription factor (TITF-2), which participates in the morphogenesis and migration of the thyroid gland. Certain polymorphisms in this gene have been more robustly associated with FNMTC. As we discussed earlier, in the 2009 GWA study by Gudmundsson et al.,30 the polymorphisms rs944289 (located at 14q13.3, near NKX2-1) and rs965513 (at 9q22.33, near FOXE1), were described as predisposing to thyroid cancer in the European population. Bonora et al.41 genotyped these and 21 other SNP identified by GWA study in 11 candidate genes in 672 subjects from 133 families with two or more members with FNMTC. They found two SNP (rs965513 and rs10759944) at 9q22.33 (near FOXE1), which were statistically consistently associated with FNMTC, although not so SNP rs944289. Pereira et al.32 then sequenced FOXE1 in 60 families with FNMTC and in 80 sporadic cases of PTC, finding a new germline variant (Ala248Gly) with segregation in a single family with FNMTC which in functional studies promoted tumourigenesis.
Therefore, although the results are not completely consistent, FOXE1 could be considered a low-penetrance FNMTC susceptibility gene.
SRGAP1 (SLIT-ROBO Rho GTPase-activating protein 1)SRGAP1 (locus 12q14) encodes a protein that inactivates CDC42, a small GTPase protein of the Rho family involved in neuronal migration and tumourigenesis.42 Through linkage studies, the 12q14 locus was identified in 21/38 families with FNMTC, and this subsequently led to the sequencing of all exons of SRGAP1 located at that level.43 They found four germline missense mutations (Gln149His, Ala275Thr, Arg617Cys and His875Arg), each in a single family. Functional studies demonstrated that the Gln149His and Arg617Cys variants caused loss of function of the SRGAP1 protein, which would be unable to inactivate CDC42. These findings suggest that SRGAP1 could be a low-penetrance gene associated with FNMTC. However, its role as a candidate gene needs to be validated in a larger cohort of families with FNMTC.
HABP2 (Hyaluronan-Binding Protein 2)HABP2 is located at 10q25.3. It encodes a protease that binds to hyaluronic acid and is involved in fibrinolysis and vascular integrity.44
The first study to apply whole-exome sequencing to study the genetic bases of FNMTC was published in 2015. Gara et al.45 described a constitutional genetic mutation Gly534Glu in the HABP2 gene, which co-segregated in a family with seven members with PTC, as a possible cause of the disease. They went on to identify this mutation in 4.7% of 423 cases with sporadic DTC, versus 0.7% of the control population, indicating a 6.71-fold higher frequency in patients with thyroid cancer. They also demonstrated overexpression of the HABP2 protein in the tumour tissue of patients with FNMTC, compared to those of controls and sporadic cases; furthermore, in the functionality studies, the presence of the Gly534Glu variant meant loss of tumour suppressor function. All this suggested that HABP2 could have a tumour suppressor function and that the Gly534Glu mutation would represent a predisposition to thyroid cancer with incomplete penetrance. Another group found 4/29 families with FNMTC that also had the same germline variant Gly534Glu.46 However, the role of HABP2 as an FNMTC susceptibility gene remains subject to debate, as these results have not been replicated in other studies and the population frequency of the Gly534Glu mutation could be highly variable depending on geographical origin.47
SRRM2 (serine/arginine repetitive matrix 2)SRRM2 (locus 16p13.3) encodes a protein involved in splicing. This gene was proposed as a candidate in FNMTC as a result of the study by Tomsic in 2015,48 in which it is suggested that variants in this protein could affect the splicing of genes involved in thyroid tumourigenesis. They performed NGS on two members of a family of six affected by FNMTC, finding the heterozygous variant c.1037C>T (Ser346Phe, rs149019598), which co-segregated with the DTC in the family. However, this mutation was not found when they analysed 138 other families with DTC and therefore requires further validation.
Genes of the telomere-telomerase complexTelomeres are repetitive sequences of non-coding DNA found at the ends of chromosomes. Their function is to provide chromosomal stability, and when these sequences are shortened, instability occurs, favouring oncogenesis.49 There are several genes involved in telomere maintenance, such as TERT (telomerase reverse transcriptase) and the genes of the shelterin complex (POT1, RAP1, TIN2, TPP1, TRF1 and TRF2).50
We know that somatic mutations in the TERT promoter play an important role in the development of thyroid cancer. It has therefore been suggested that genes involved in telomere maintenance could be involved in DTC, and in FNMTC in particular. In fact, patients with FNMTC have been described as having shorter telomeres than their unaffected relatives, healthy controls or sporadic cases.51 However, Jendrzejewski et al.52 found no difference in telomere length between sporadic DTC and FNMTC. Nor were any differences found in the number of copies or in the expression of various genes of the telomere-telomerase complex in six families with FNMTC,53 and no germline mutations were found in TERT or other shelterin complex genes in families with FNMTC.50,54 Therefore, the results in the literature are still fairly inconsistent and further research is required.
RTFC or C14orf93 (chromosome 14 open reading frame 93)A 2017 study in China55 that combined linkage analysis and NGS found the variant Val205Met in RTFC (locus 14q11.2) in a single family with five members with FNMTC. In functional studies, this variant promoted oncogenesis. These results have not been validated in other independent studies.
MAP2K5 (mitogen-activated protein kinase kinase 5)MAP2K5 (also known as MEK5), is located at 15q23 and encodes a protein from the MAP kinase family. A recent article on the Chinese population56 studied 34 families with three or more members affected by FNMTC using NGS, and found two variants in the MAP2K5 gene present in two families: Ala321Thr and Met367Thr. These variants are very uncommon in the healthy population. The results of functional studies also suggest that MAP2K5 could be an FNMTC susceptibility gene. However, another Italian group did not replicate these results in 33 families,57 and so the involvement of MAP2K5 in this disease remains uncertain.
DUOX2 (dual oxidase 2)Located at 15q21.1, this gene encodes a glycoprotein that participates in the metabolism of hydrogen peroxide (H202), necessary for the activity of thyroid enzymes involved in the synthesis of thyroid hormones. A new germline variant has recently been described in DUOX2 (the missense variant Tyr1203His), identified by NGS in a family with six members with FNMTC, which could be associated with susceptibility to FNMTC.58 The presence of this variant would increase the production of hydrogen peroxide, toxic to DNA, promoting tumourigenesis. Interestingly, the expression of DUOX2 is increased in patients homozygous for the polymorphism rs965513 in FOXE1, meaning both mechanisms could be connected, with the authors suggesting that the dysregulation of proteins involved in the metabolism of H2O2 could be related to susceptibility to FNMTC.
NOP53 (ribosome biogenesis factor)NOP53 is located at 19q13.33 and encodes a nucleolar protein involved in ribosomal biogenesis, regulating the activation of p53.59 It is involved in the PI3K/AKT signalling pathway, one of the most important in thyroid cancer. In fact, in a study of a family with three members with FNMTC using NGS, three susceptibility genes (PIK3CB, CAV2 and KANK1) were discovered associated with tumourigenesis via the PI3K/AKT pathway.60
Following a multicentre study of 45 families with FNMTC conducted in Spain, NOP53 has been described as another candidate gene in FNMTC. The study found the Asp31His variant (rs78530808), which co-segregated in all affected members of three families with five, four and two members suffering from DTC, respectively, and was not present in healthy relatives. Furthermore, functional studies showed NOP53 to have an oncogenic function.61 In fact, in a previous study, greater expression of NOP53 had been detected in aggressive follicular carcinomas.62
NOP53 could therefore be involved in FNMTC, probably as a low-penetrance gene, although these recent findings have not yet been corroborated in other studies.
MicroRNAIt has also been hypothesised that microRNA, small RNA segments capable of regulating the expression of other genes, could be involved in the development of thyroid cancer. A 2018 review63 discusses the different microRNA associated at that time with the different subtypes of thyroid cancer. The article by Xiong et al.64 is worthy of note as it focusses on the study of the microRNA profile in FNMTC, finding a dysregulation of miR-886-3p and miR-20-a.
LimitationsAll of the above serves to demonstrate the complexity of the study of the genetics of FNMTC for various reasons.
First of all, the difficulty of gathering clinical data and biological samples (blood and tumour histological tissue) from families with several affected cases, as multicentre collaboration is required. In fact, the Spanish Society of Endocrinology and Nutrition (SEEN) has a registry in which data pertaining to cases of FNMTC we come across in our clinical practice can be input (http://www.regcancerdiferenciadofamiliar.es/). Based on the studies cited above, it seems that our efforts should focus on families with three or more affected members, in order to maximise the hereditary component. As we said earlier, some characteristics such as tumour aggressiveness and younger age at diagnosis, or the utility of screening, are much clearer in families with at least three affected people. It therefore seems better to prioritise the homogeneity and quality, rather than the quantity, of the families recruited. Furthermore, given the high prevalence of thyroid cancer in the general population, it should not be ruled out that within a family with FNMTC there may be a case of sporadic DTC or, in other words, a phenocopy.
Secondly, considering the inconsistency of results between one study and another, which essentially suggest low-penetrance candidate genes or even single mutations in some families, unlike most hereditary cancers, which usually have one or two causative genes, perhaps FNMTC could be a genetically heterogeneous and polygenic disease. This would explain why no common link has been found between the different families.
Lastly, although whole-exome sequencing techniques have represented a great qualitative leap in biochemistry and molecular genetics, and are increasingly standardised, they still have their limitations and frequently require validation by Sanger sequencing. Moreover, the results and variants of interest obtained depend to a great extent on the search strategy and filters used. These elements are essential due to the enormous volume of information available, but at the same time create a selection bias, as we may miss potential candidate variants that cannot be completely ruled out.65
ConclusionsFNMTC is still a heterogeneous disease with no established genetic cause. Except in the case of FNMTC associated with hereditary genetic syndromes, genetic study of families with FNMTC is not indicated, as no genes have been clearly established as causative. Nevertheless, it is a good idea to collect data and register these families, to enable multicentre collaborative studies to be carried out to help identify the genetic causes of their disease.
FundingThis study received no funding of any kind.
Conflicts of interestThe authors declare that they have no conflicts of interest.
Please cite this article as: Orois A, Mora M, Halperin I, Oriola J. Carcinoma diferenciado de tiroides familiar: más allá de las formas sindrómicas. Endocrinol Diabetes Nutr. 2021;68:260–269.
