




El tumor neuroectodérmico maligno del tracto gastrointestinal (GNET) es una neoplasia maligna sumamente rara, descrita por primera vez por Zambrano et al. en 2003 como tumor similar al sarcoma de células claras del tracto gastrointestinal, pues a diferencia del sarcoma de células claras posee células gigantes osteoclásticas y positividad difusa e intensa para S-100 con ausencia inmunohistoquímica y ultraestructural de diferenciación melanocítica. La presente publicación busca aportar los 2 primeros casos de GNET reportados en nuestro país, Perú, y América Latina. Reportamos 2 casos de GNET, en paciente mujer y en varón, ambos en la sexta década de la vida, cuyos casos llegaron a nuestra institución para reevaluación diagnóstica. Uno de los casos continuó tratamiento en nuestro instituto con evolución desfavorable. El conocimiento de su existencia y criterios diagnósticos por los patólogos es necesario para evitar confundirla y maldiagnosticarla con alguna otra neoplasia gastrointestinal no epitelial.
Malignant gastrointestinal neuroectodermal tumour (GNET) is an extremely rare neoplasm first described by Zambrano in 2003 as clear cell sarcoma like tumor of the gastrointestinal tract. In contrast to clear cell sarcoma, it has giant osteoclast cells and shows diffuse and intense positivity for S-100 with no immunohistochemical or ultrastructural melanocyte differentiation. We present the first cases of GNET reported in South America, occurring in Peru. Two cases of GNET, one in a female and one in a male, both between 60 and 70 years of age, were referred to our hospital for reevaluation. One underwent further treatment in our centre, but with an unfavourable evolution. Pathologists should be aware of the diagnostic criteria for GNET in order to avoid misdiagnosis due to confusion with other non-epithelial gastrointestinal neoplasms.
El tumor neuroectodérmico maligno del tracto gastrointestinal (GNET) es una neoplasia maligna sumamente rara que se origina dentro de la pared del intestino delgado, estómago o intestino grueso y el cual, además, se caracteriza por presentar células gigantes multinucleadas tipo osteoclastos, lo que recuerda al sarcoma de células claras (CCS) de partes blandas, por eso inicialmente, e incluso en algunos casos se le sigue denominando como tumor similar al CCS del tracto gastrointestinal (CCSLTGT)1–3.
Fue descrito por primera vez por Zambrano et al. en 2003, quienes reportaron una serie de 6 casos de tumores que recordaban, pero que a la vez diferían de los CCS de tejidos blandos por poseer células gigantes osteoclásticas, ausencia de pigmento de melanina en las células tumorales y positividad intensa y difusa para S-100 con ausencia inmunohistoquímica y ultraestructural de diferenciación melanocítica4. Pese a compartir la traslocación del EWSR1 con el CCS de partes blandas, hoy por hoy se sabe que se tratan de entidades distintas e independientes.
Actualmente, podemos encontrar 91 casos reportados de GNET, ya sea con las nomenclaturas de GNET, CCSLTGT o CCS del tracto gastrointestinal (TGI) pero con falta de diferenciación melanocítica2,3,5,6, todos los cuales provienen de literatura inglesa, europea o asiática, sin contar con aportes de América Latina. Es por ello que en la presente publicación aportamos los 2 primeros casos de GNET reportados en nuestro país, Perú, y América Latina.
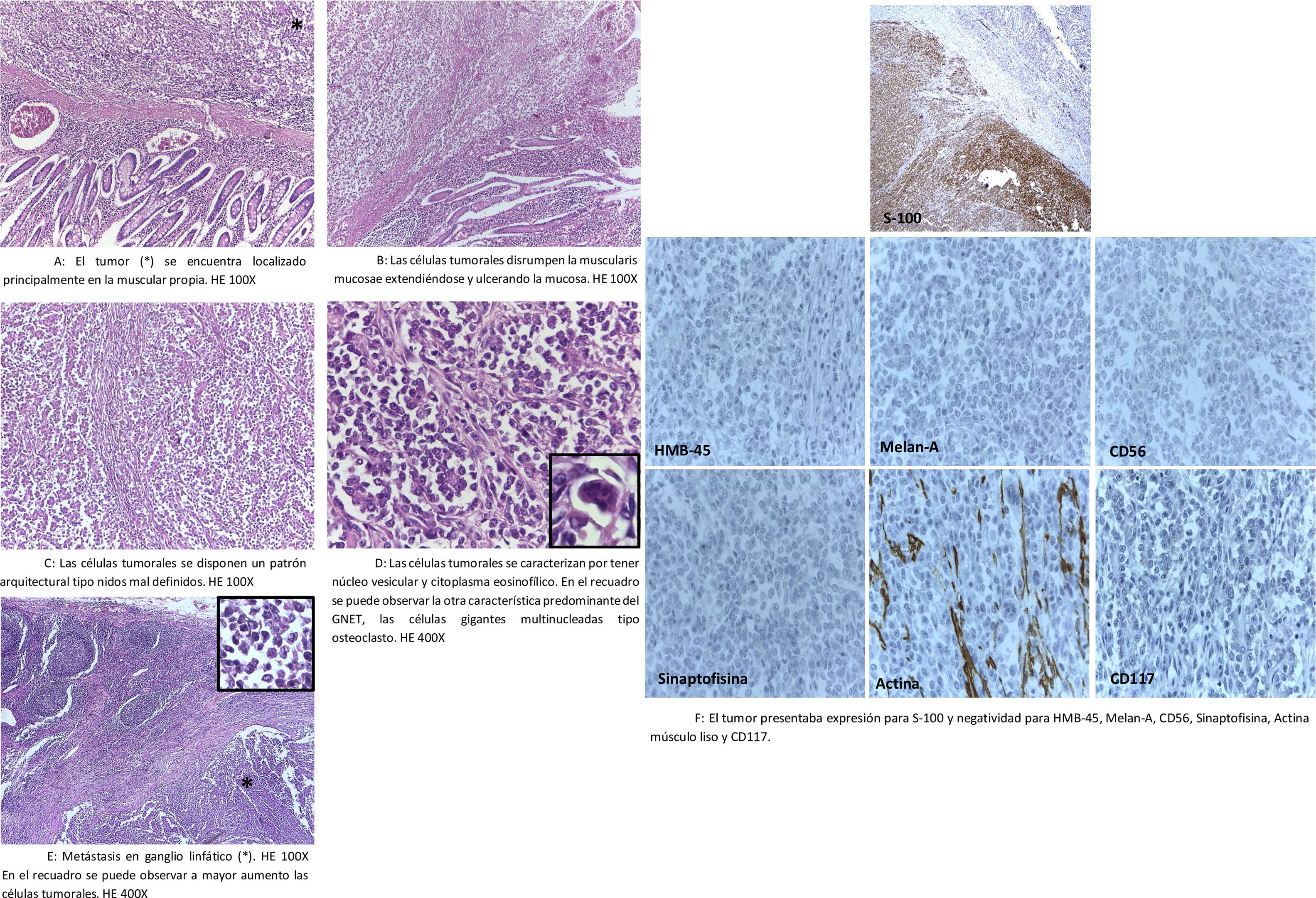
Descripción de casos clínicosPaciente 1Paciente mujer de 53 años con antecedente en el 2005 de cáncer de mama izquierda operada y con tratamiento quimioterápico incompleto. En 2011, presenta episodios de sangrado digestivo asociado a tumoración yeyunal, por lo que es sometida a resección intestinal en otra institución, con resultado anatomopatológico de sarcoma neurogénico, luego de lo cual recibe 4 cursos de quimioterapia y es referida a nuestro instituto para continuación de tratamiento. Al ingreso, el examen físico fue normal y los controles imagenológicos a los 3 meses poscirugía no revelaban metástasis ni enfermedad residual. La lectura e informe de los bloques y láminas de inmunohistoquímica, procedentes de la otra institución, concluyeron el diagnóstico de tumor del TGI rico en osteoclastos y con características similares a CCS y macrometástasis ganglionar en 2 de 4 ganglios linfáticos (fig. 1A-E). El inmunofenotipo del tumor presentaba expresión para S-100 y vimentina; mientras que negatividad para melan-A, HMB-45, CD117, CD34, CD56, panqueratina, ACL, CD3 y CD20 (fig. 1F). Asimismo, se pudo confirmar por FISH la presencia de la traslocación del gen EWSR1 (22q12), realizado en Mayo Clinic, Rochester Minessota-EE. UU.
 se encuentra localizado principalmente en la muscular propia. HE 100X. B: Las células tumorales disrumpen la muscularis mucosae, extendiéndose y ulcerando la mucosa. HE 100X. C: Las células tumorales se disponen en un patrón arquitectural tipo nidos mal definidos. HE 100X. D: Las células tumorales se caracterizan por tener núcleo vesicular y citoplasma eosinofílico. En el recuadro se puede observar la otra característica predominante del GNET, las células gigantes multinucleadas tipo osteoclasto. HE 400X. E: Metástasis en ganglio linfático (*). HE 100X. En el recuadro se puede observar a mayor aumento las células tumorales. HE 400X. F: El tumor presentaba expresión para S-100 y negatividad para HMB-45, melan-A, CD56, sinaptofisina, actina músculo liso y CD117.")
A: El tumor (*) se encuentra localizado principalmente en la muscular propia. HE 100X. B: Las células tumorales disrumpen la muscularis mucosae, extendiéndose y ulcerando la mucosa. HE 100X. C: Las células tumorales se disponen en un patrón arquitectural tipo nidos mal definidos. HE 100X. D: Las células tumorales se caracterizan por tener núcleo vesicular y citoplasma eosinofílico. En el recuadro se puede observar la otra característica predominante del GNET, las células gigantes multinucleadas tipo osteoclasto. HE 400X. E: Metástasis en ganglio linfático (*). HE 100X. En el recuadro se puede observar a mayor aumento las células tumorales. HE 400X. F: El tumor presentaba expresión para S-100 y negatividad para HMB-45, melan-A, CD56, sinaptofisina, actina músculo liso y CD117.
Los continuos controles tomográficos y de RMN poscirugía continúan siendo negativos por un lapso de un año y 2 meses, luego de lo cual la paciente es perdida de vista hasta enero del 2014, reingresando a nuestra institución por presentar dolor abdominal, pérdida de peso, melenas intermitentes y deterioro progresivo. Es sometida a laparotomía exploratoria, con la cual en segmentos hepáticos 4 y 8 se encuentran 2 implantes nodulares, así como tumoración retroperitoneal ganglionar de 8×8×6cm hacia el lado izquierdo, que engloba vasos mesentéricos superiores con compromiso de la vasculatura cólica media y que llega hasta la vena mesentérica inferior, adhiriéndose, además, a la pared posterior gástrica, por todo ello es imposible resecarla. Debido a la evidencia de recurrencia masiva tumoral asociada a metástasis hepática no es tributaria a tratamiento quirúrgico, ni adyuvante con radioterapia ni quimioterapia; por lo que es dada de alta con medidas de sostén y pronóstico malo a corto plazo.
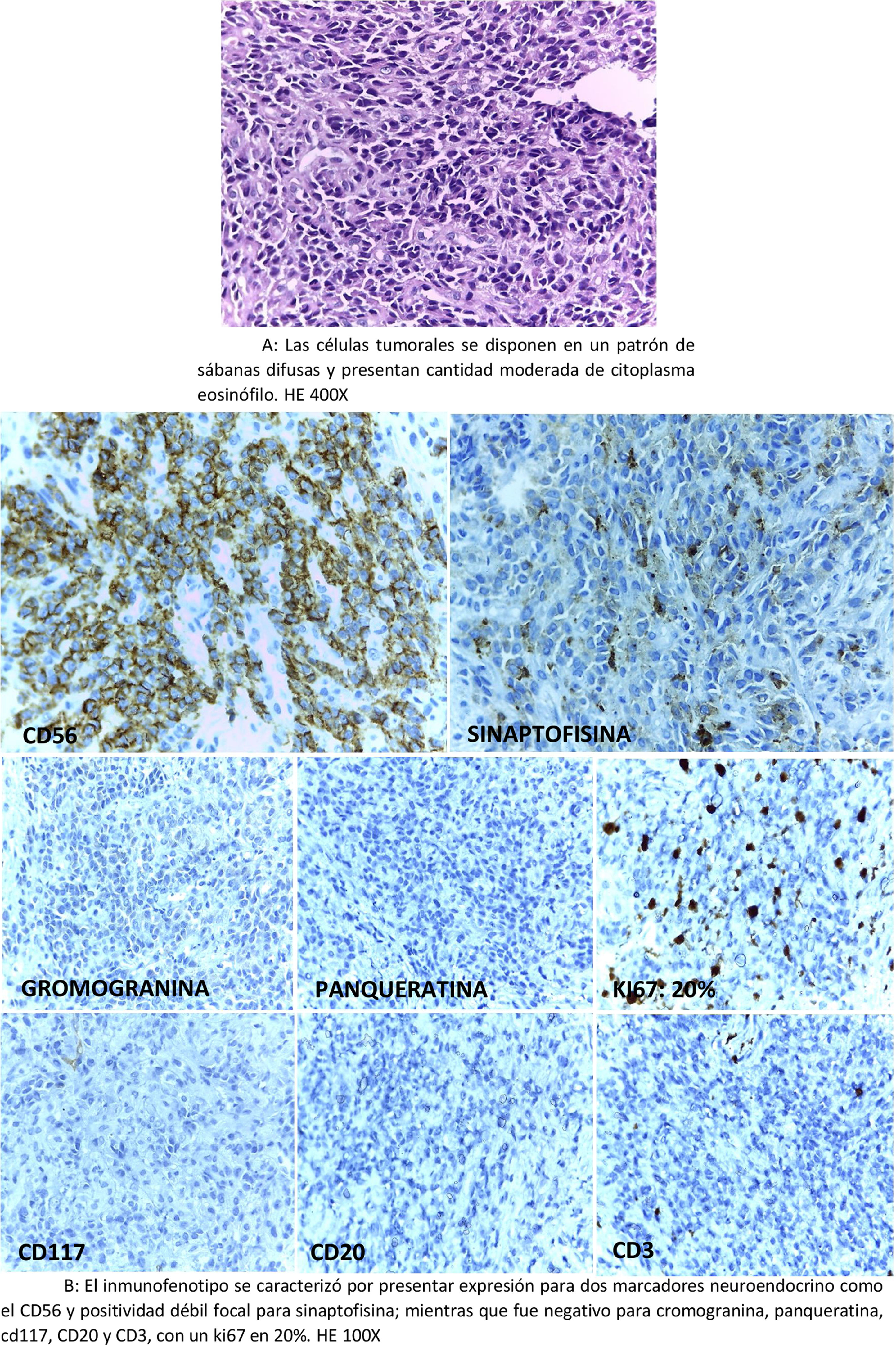
Paciente 2Paciente varón de 52 años con antecedente de carcinoma neuroendocrino G3, diagnóstico realizado en abril del 2018 por medio de biopsia colónica, al presentar, este tumor, una morfología de alto grado y patrón arquitectural difuso (fig. 2A); asimismo, el inmunofenotipo expresaba positividad difusa e intensa para CD56 y débil y focal para sinaptofisina con un ki67 en 20%; mientras que fue negativo para cromogranina, CD117, CD20, CD3 y panqueratina (fig. 2B).

A: Las células tumorales se disponen en un patrón de sábanas difusas y presentan cantidad moderada de citoplasma eosinófilo. HE 400X. B: El inmunofenotipo se caracterizó por presentar expresión para 2 marcadores neuroendocrino como el CD56 y positividad débil focal para sinaptofisina; mientras que fue negativo para cromogranina, panqueratina, cd117, CD20 y CD3, con un ki67 en 20%. HE 100X.
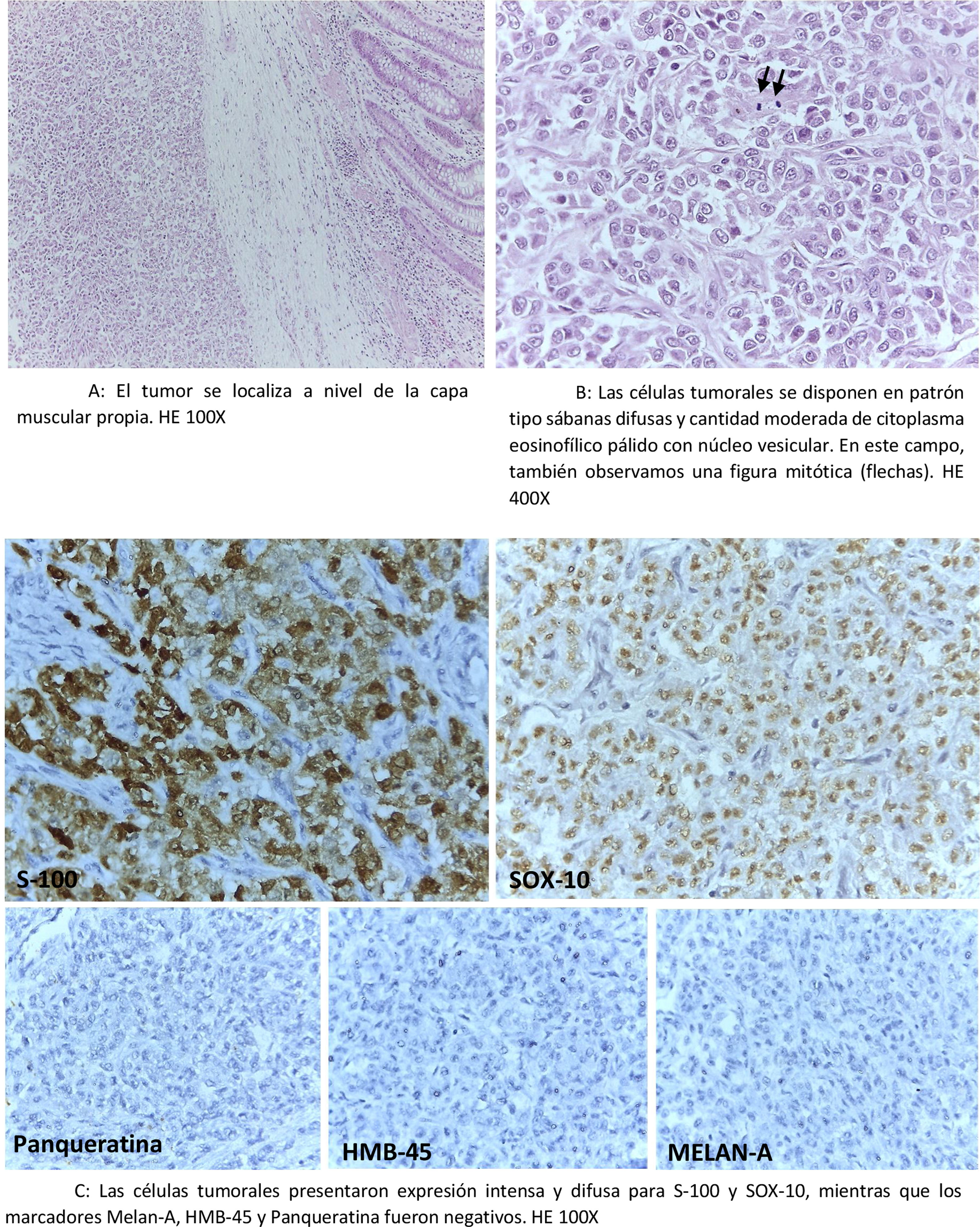
Posteriormente, en agosto del 2018 es sometido a hemicolectomía derecha en otra institución, cuyos bloques de parafina son remitidos a nuestro instituto para una segunda evaluación y mayor precisión del diagnóstico patológico. La lectura y evaluación de los mismos, sumado a la reevaluación de las láminas de la biopsia colonoscópica inicial concluyeron el diagnóstico de GNET, puesto que el inmunofenotipo, además de lo encontrado en la biopsia quirúrgica reveló positividad para S-100 y SOX-10, mientras que los marcadores melan-A, HMB-45 y panqueratina fueron negativos (fig. 3A-C).
. HE 400X. C: Las células tumorales presentaron expresión intensa y difusa para S-100 y SOX-10, mientras que los marcadores melan-A, HMB-45 y panqueratina fueron negativos. HE 100X.")
A: El tumor se localiza a nivel de la capa muscular propia. HE 100X. B: Las células tumorales se disponen en patrón tipo sábanas difusas y cantidad moderada de citoplasma eosinofílico pálido con núcleo vesicular. En este campo, también observamos una figura mitótica (flechas). HE 400X. C: Las células tumorales presentaron expresión intensa y difusa para S-100 y SOX-10, mientras que los marcadores melan-A, HMB-45 y panqueratina fueron negativos. HE 100X.
El GNET es una neoplasia maligna extremadamente rara, la cual desde su primera descripción con la denominación de CCSLTGT por Zambrano et al. en 20033,4 se ha considerado como una entidad independiente, pues a diferencia de los CCS de partes blandas carece de diferenciación melanocítica desde el punto de vista morfológico, inmunofenotípica y ultraestructuralmente. En 2012, Stockman et al.7 propusieron una nueva nomenclatura para este tumor, a GNET, término que sigue adquiriendo cada vez mayor aceptación. Actualmente, encontramos 91 casos reportados, ya sea con las denominaciones de GNET, CCSLTGT o CCS del TGI pero con falta de diferenciación melanocítica2,3,5,6, todos los cuales provienen de literatura inglesa, europea o asiática, lo que ha permitido caracterizar al tumor desde el punto de vista clínico, histopatológico y genético. La presente publicación busca aportar los 2 primeros casos de GNET reportados en nuestro país, Perú, y América Latina.
El GNET tiende a ocurrir en adultos jóvenes, con una edad media de 35 años y no existe predominio por el sexo. Se le ha descrito exclusivamente dentro de la cavidad abdominal, la mayoría originándose en la pared del intestino delgado, incluyendo el íleon y yeyuno, pero también se ha documentado en el estómago, colon y peritoneo. La principal sintomatología es el dolor abdominal con obstrucción intestinal1–4,7–10.
La etiología sigue siendo desconocida. No obstante, existen 2 reportes de GNET en pacientes con antecedente de hepatoblastoma y sarcoma de Ewing durante la infancia, sugiriendo que la anomalía genética en el estadio de embriogénesis puede ser un factor de riesgo para la oncogénesis de este tumor. Asimismo, existe un reporte en el que el GNET ocurrió como neoplasia secundaria a radiación, en un paciente con historia de neuroblastoma en la niñez, proponiendo que la radioterapia también podría estar involucrada en el desarrollo de este tumor en los próximos años de vida1–3,11–13.
Macroscópicamente, se centran dentro de la capa muscular propia, pudiendo, a menudo, extenderse a la submucosa y subserosa. Algunos tumores crecen como una masa polipoide, que se extiende dentro de la luz con la consecuente ulceración de la mucosa, mientras que otros se presentan como lesiones estenosantes circunferenciales. Alcanzan un tamaño promedio de 4,5cm y la superficie de corte tiene un aspecto sólido con bordes multinodulares e infiltrativos, color pardo blanquecino con cantidades variables de hemorragia y necrosis e incluso cambios quísticos1,3,4,6,7.
Histológicamente, son tumores con alta celularidad e infiltración difusa que tienden a obliterar la pared intestinal o gástrica, con ulceración de la mucosa y extensión a la serosa. Arquitecturalmente, el patrón más clásico que adopta es de agrupaciones tipo sábanas difusas o nidos de borden mal definidos, característica que lo diferencia de los CCS que, por el contrario, adoptan un patrón de nidos con bordes bien delimitados. A menudo, los GNET también pueden presentar, e incluso focalmente, áreas con otros patrones de crecimiento como el seudoalveolar, seudopapilar, microquístico, fascicular, trabecular o formar estructuras tipo rosetas.
Las células tumorales suelen ser monomórficas de apariencia epitelioide a ovoides medianas a relativamente grandes con cantidad variable de citoplasma eosinófilo pálido y, menos frecuente, citoplasma completamente claro. El núcleo es central, poligonal con cromatina vesicular y nucléolo a menudo pequeño e inconspicuo. La actividad mitótica es variable con rangos entre 0 a 20 mitosis por campo de gran aumento (400X). Otra característica relevante, y que lo distingue de otras neoplasias de células claras, es la presencia de células gigantes multinucleadas tipo osteoclasto CD68 positivo, cuyo número varía marcadamente entre tumores o incluso entre uno y otro campo dentro de la misma tumoración; aunque, existen algunos casos que no las presentan. Finalmente, los GNET carecen de pigmento melánico en todos los casos reportados hasta la actualidad, lo que favorece su distinción de los melanomas y CCS1–4,6. Las recurrencias o metástasis retienen la morfología del tumor primario incluyendo las células gigantes multinucleadas tipo osteoclasto; no obstante, las células tumorales pueden presentar mayor grado de pleomorfismo1,7.
Inmunohistoquímicamente tienen como característica indistinguible la expresión de la proteína S-100, que varía de difusa e intensa, en la mayoría de los casos, a parcheada; acompañada de la ausencia de expresión de marcadores melanocíticos específicos como HMB-45, melan-A, tirosina y MiTF. Asimismo, expresan SOX-10, que es un factor transcripcional responsable del desarrollo de la cresta neural. Este patrón inmunofenotípico indica que los GNET se originan de células precursoras neuroectodérmicas que son incapaces de diferenciarse hacia un linaje melanocítico3,6.
Existe hoy en día mayor evidencia de que los GNET presentan reactividad variable para al menos un marcador neuroendocrino como CD56, sinaptofisina y/o neuroenolasa específica1–3,6,7, por lo que ante biopsias de tumores del TGI que no muestren una diferenciación neuroendocrina concluyente, tal como lo ocurrido en la biopsia inicial del caso 2 que reportamos, se debe descartar que no se trate de un GNET mediante el uso de los marcadores inmunohistoquímicos previamente descritos. En contraparte, estos tumores son negativos para marcadores asociados a tumor estromal gastrointestinal, desmina, actina músculo liso, marcadores epiteliales y CD99, lo que permite diferenciarlo de sus principales diagnósticos diferenciales como el CCS del TGI, melanoma, tumor estromal gastrointestinal, sarcoma sinovial, tumor maligno de la vaina del nervio periférico, entre otros1–3.
Estudios de microscopia electrónica describen las células tumorales como poligonales con múltiples procesos celulares interdigitantes en adición a gránulos secretores densos, consistente con diferenciación neuroendocrina, sin presencia de melanosomas o estructuras tipo melanosomas, lo que avala lo encontrado en la expresión inmunofenotípica y el porqué el nombre más apropiado es GNET1–4,7. Genéticamente, presentan alteraciones del gen EWSR1 en la mayoría de los casos investigados, que incluyen fusiones de genes EWSR-CREB y EWSR-ATF; sin embargo, traslocaciones del gen EWSR1 se han detectado en otros tumores, incluyendo CCS o algunos carcinomas neuroendocrinos; por lo que la presencia de traslocación del gen EWSR1 no es un criterio específico para GNET, pero sí ayuda a confirmar el diagnóstico de este2,3,5,6,14.
Finalmente, en cuanto al pronóstico, la presencia de metástasis al momento del diagnóstico es frecuente, principalmente a hígado y ganglios linfáticos. La media de supervivencia suele ser de 18,5 meses, con un rango entre 3 a 106 meses1–7. El manejo usual consiste en la resección del segmento de intestino comprometido, seguido de controles de seguimiento rigurosos mediante estudios de imágenes para detectar recurrencia y/o metástasis. Y aunque la literatura reporta casos que recibieron quimioterapia poscirugía, esta aún no se considera dentro del protocolo terapéutico1,3,9,12.
En conclusión, los GNET son tumores malignos extremadamente raros del TGI, con solo 93 casos hasta la actualidad, incluyendo los 2 casos que presentamos en esta publicación, los cuales, además, constituyen los primeros registrados en nuestro país, Perú, y América Latina2,3,5,6. El conocimiento de su existencia y criterios diagnósticos por los patólogos es necesario para evitar confundirla y maldiagnosticarla con alguna otra neoplasia GI no epitelial.
Conflicto de interesesLas autoras declaran que no existen conflictos de intereses respecto a la publicación del presente reporte de caso.
Las autoras queremos expresar nuestro reconocimiento al Dr. Andre M. Oliveira del Departamento de Patología de la Clínica Mayo, Rochester Minnesota-EE. UU. por su contribución en el estudio molecular por FISH que se realizó en el caso n.o 1 que reportamos.

