





El síndrome de Sweet o dermatosis neutrofílica febril aguda es una enfermedad inflamatoria infrecuente de fisiopatología desconocida, aunque las evidencias clínicas y bioquímicas sugieren que las citocinas tienen un papel importante en su etiopatogenia. Se distinguen 5 grupos etiológicos: idiopático, parainflamatorio, secundario a fármacos, asociado a embarazo y paraneoplásico. Este último grupo representa el 20% de los casos, asociados el 85% de ellos a neoplasias hematológicas y el 15% a tumores sólidos.
PacienteSe presenta el caso de un paciente afecto de síndrome de Sweet con afectación dermatológica atípica, asociado a un síndrome mielodisplásico y también a un síndrome de Cushing iatrogénico secundario a altas dosis de corticoides cuya evolución fue desfavorable.
ResultadosPositividad de los reactantes de fase aguda (RFA) durante los brotes, destacando la elevación precoz de la interleucina 6 (IL-6) seguida del seroamiloide A (SAA) y proteína C reactiva (PCR), encontrándose diferencias estadísticamente significativas (p<0,05) entre la PCR y el SAA.
ConclusionesUn síndrome de Sweet en un varón con múltiples recaídas y localización dermatológica no clásica, cuando se asocia a alteraciones hematológicas, con un incremento de los RFA y elevación precoz de la IL-6 debe orientar el diagnóstico clínico hacia un origen paraneoplásico y hematológico.
Sweet's syndrome or acute febrile neutrophilic dermatosis is a rare inflammatory disease of unknown pathophysiology, although clinical and biochemical evidence suggests that cytokines play an important role in its aetiopathogenesis. It is classified into five groups: idiopathic, para-inflammatory, secondary to drugs, associated with pregnancy, and para-neoplastic in 20% of cases, with 85% of these linked to haematological disorders, and 15% to solid tumours.
PatientA report is presented on a patient with Sweet's Syndrome with atypical dermatological involvement, associated with myelodysplastic syndrome, and iatrogenic Cushing's syndrome secondary to high-doses of corticosteroids, with an unfavorable outcome.
ResultsAcute phase reactants (APR) were increased during the outbreaks, with the early elevation of interleukin 6 (IL6) being highlighted, followed by serum amyloid A (SAA) and C-reactive protein (CRP), with statistically significant differences (P<.05) between CRP and SAA.
ConclusionsA Sweet's syndrome in a male with multiple relapses and a non-classical dermatological location, associated haematological abnormalities, and an increase in APR with early elevation of IL6, should lead to a clinical diagnosis of paraneoplastic and haematological origin.
El síndrome de Sweet (SS) es una enfermedad inflamatoria poco frecuente que se caracteriza porque el paciente presenta fiebre, leucocitosis y placas eritematosas dolorosas de comienzo brusco. El estudio anatomopatológico demuestra la presencia de densos infiltrados de neutrófilos en la dermis papilar sin vasculitis1,2. Su y Liu propusieron los criterios diagnósticos de esta enfermedad actualmente vigentes3 con las modificaciones realizadas por Von der Driesch et al.4. Aunque la fisiopatología es desconocida, el mecanismo etiopatogénico se le atribuye a un trastorno en la regulación de las citocinas2,5,6, siendo los corticoides sistémicos en la actualidad el gold standard del tratamiento7,8.
El SS puede clasificarse en 5 grupos: idiopático, parainflamatorio, paraneoplásico, secundario a fármacos y asociado al embarazo. En el 20% de los casos se asocia a las enfermedades malignas representando las hematológicas el 85% y los tumores sólidos el 15% restante8. Con respecto a las neoplasias hematológicas la leucemia mieloide aguda es la más frecuente (42%), seguida de los linfomas (11%), los síndromes mielodisplásicos (SMD) (9%) y la leucemia mielocítica crónica (7%)2. Las mielodisplasias son un grupo heterogéneo de trastornos hematológicos que se caracterizan por presentar citopenias debidas a los estados dismórficos de las células de la médula ósea1.
Se expone el caso de un paciente diagnosticado de un síndrome mielodisplásico multilínea de riesgo intermedio, con un SS paraneoplásico asociado y complicado con un síndrome de Cushing iatrogénico, después de más de 3 años de tratamiento con altas dosis de corticoides orales y parenterales en los ingresos hospitalarios, que evolucionó de forma tórpida y recurrente ocasionándole la muerte.
PacienteEl paciente es un varón de 47 años que consulta por presentar un cuadro de malestar general, fiebre de 39°C con escalofríos, astenia, mialgias y artralgias, precedido en los días previos de tos con expectoración blanquecina escasa. A las pocas horas de instaurarse la fiebre apareció un exantema doloroso de distribución universal, con predominio en las extremidades inferiores en forma de placas de bordes sobreelevados, calientes y muy dolorosas que fueron etiquetadas con el diagnóstico de eritema nodoso y eritema multiforme. El tratamiento indicado fue reposo, claritromicina y diclofenaco, que fue sustituido por indometacina, que resultó mucho más efectivo, experimentando el paciente una leve mejoría. A las 3 semanas de su recuperación sufrió un nuevo brote evolucionando con una clínica muy similar, pero se acentuó la sintomatología respiratoria presentando tos, expectoración y disnea. Se aplicó el mismo tratamiento, pero los antiinflamatorios no esteroideos (AINE) fueron sustituidos por deflazacort, observando una rápida mejoría. En los brotes sucesivos la debilidad muscular fue cada vez más extrema, con fiebre, sudoración profusa, disnea intensa y con la implicación de otros órganos además del pulmón, encéfalo, bronquios, intestino, hígado, ojos (epiescleritis) y aparato genitourinario (uretritis) en el contexto sistémico descrito. En la figura 1 se describe el cuadro clínico del paciente durante su enfermedad. Se realizó el diagnóstico diferencial con las enfermedades reumatológicas del tipo artritis reumatoide, dermatomiositis, artritis reactivas, granulomatosis de Wegener o la enfermedad de Behçet, entre otras, así como las trombofilias genéticas y adquiridas como el síndrome antifosfolípido.

En el ingreso debido al cuarto brote se realizaron varias biopsias a diferentes niveles: en la piel se describieron unas lesiones histopatológicas compatibles con el SS, en la del músculo cuádriceps no se presentan signos relevantes, mientras que en la médula ósea se encuentra una hiperplasia total con dishemopoyesis global y el 5% de blastos y un SMD tipo anemia refractaria con un exceso de blastos (AREB). En la ecocardiografía se detectó una hipertensión pulmonar. El diagnóstico establecido fue de SS y se instauró un tratamiento con corticoides sistémicos a dosis crecientes en cada brote, no recidivando las lesiones dérmicas, que solo aparecieron en los 2 primeros brotes. La mielodisplasia fue tratada con 5 azacitidina.
En los sucesivos brotes, la sintomatología respiratoria se fue agravando precisando el ingreso en la unidad de cuidados intensivos (UCI) en 2 ocasiones, así como ventilación mecánica. El cultivo microbiológico del aspirado bronquial y lavado broncoalveolar fue negativo. Desde el último ingreso en la UCI el paciente presentó un estado de debilidad generalizada, permaneciendo en cama, en tratamiento con altas dosis de deflazacort, fentanilo transdérmico y oxigenoterapia continua, produciéndose el éxitus después de 4 años de enfermedad.
ResultadosEl paciente fue diagnosticado de SS de acuerdo a los criterios diagnósticos establecidos por Su y Liu3 en la tabla 1. Además de los 2 criterios mayores, de obligado cumplimiento, como criterios menores presentó fiebre, que precedió a un cuadro de artralgias y mialgias en el contexto de un SMD con buena respuesta a corticoides sistémicos. La duración media de los brotes osciló entre 8 y 24 semanas, durante los mismos se produce una alteración de los datos biológicos. Como consecuencia del SMD se observa una anemia macrocítica progresiva, con valores de ácido fólico y vitamina B12 dentro del intervalo de referencia, trombocitopenia y leucopenia, observándose una inversión de la fórmula con predominio linfomonocítico en los rebrotes.
Criterios diagnósticos de síndrome de Sweet
| Criterios mayores | Criterios menores |
|---|---|
| - Comienzo abrupto de placas o nódulos eritematosos o violáceos y dolorosos - Infiltración dérmica de neutrófilos polimorfonucleares sin vasculitis leucocitoclástica | - Pródromos de fiebre o de proceso infeccioso - Leucocitosis - Asociación de artralgias, conjuntivitis, fiebre o neoplasia subyacente - Buena respuesta a corticoides sistémicos, pero no a la antibioterapia |
Diagnóstico: 2 criterios mayores y 2 menores.
Fuente: modificado de Su y Liu3.
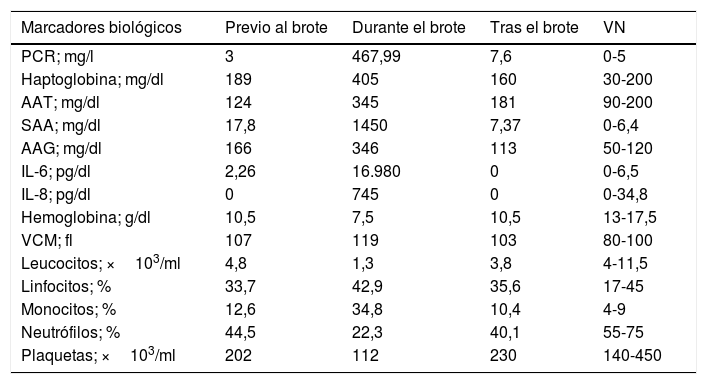
Desde la perspectiva bioquímica todos los reactantes de fase aguda eran positivos: seroamiloide A (SAA), alfa-1 antitripsina (AAT), orosomucoide o alfa-1 glicoproteína ácida (AAG) y la haptoglobina (tabla 2), siendo las interleucinas (IL) 6, 8, PCR y SAA las que se elevan más precozmente (fig. 2).
Valores de los reactantes de fase aguda, interleucinas y parámetros hematológicos durante la evolución de un brote
| Marcadores biológicos | Previo al brote | Durante el brote | Tras el brote | VN |
|---|---|---|---|---|
| PCR; mg/l | 3 | 467,99 | 7,6 | 0-5 |
| Haptoglobina; mg/dl | 189 | 405 | 160 | 30-200 |
| AAT; mg/dl | 124 | 345 | 181 | 90-200 |
| SAA; mg/dl | 17,8 | 1450 | 7,37 | 0-6,4 |
| AAG; mg/dl | 166 | 346 | 113 | 50-120 |
| IL-6; pg/dl | 2,26 | 16.980 | 0 | 0-6,5 |
| IL-8; pg/dl | 0 | 745 | 0 | 0-34,8 |
| Hemoglobina; g/dl | 10,5 | 7,5 | 10,5 | 13-17,5 |
| VCM; fl | 107 | 119 | 103 | 80-100 |
| Leucocitos; ×103/ml | 4,8 | 1,3 | 3,8 | 4-11,5 |
| Linfocitos; % | 33,7 | 42,9 | 35,6 | 17-45 |
| Monocitos; % | 12,6 | 34,8 | 10,4 | 4-9 |
| Neutrófilos; % | 44,5 | 22,3 | 40,1 | 55-75 |
| Plaquetas; ×103/ml | 202 | 112 | 230 | 140-450 |
AAG: alfa-1 glicoproteína ácida; AAT: alfa-1 antitripsina; IL: interleucina; PCR: proteína C reactiva; SAA: seroamiloide A; VN: valores normales.

Todos los datos biológicos recogidos durante los brotes y periodos interbrotes fueron analizados mediante los programas informáticos R versión 3.0.2 (2013-09-25) - Frisbee Sailing IBM® SPSS Statistics® v. 22, ed. 64 bits. Se estudia la relación entre las variables por medio de la correlación de Pearson y se aplica el test de independencia asociado, encontrándose una relación lineal entre el número de plaquetas y la concentración de hemoglobina y también entre la PCR con la IL-6, SAA, AAT, haptoglobina y AAG, así como entre la AAT en relación con la AAG, SAA, PCR y haptoglobina, encontrándose diferencias estadísticamente significativas (p<0,05) entre los valores de PCR y SAA durante los brotes en relación con los periodos interbrote. Destaca también la negatividad de las pruebas de autoinmunidad y la hipogammaglobulinemia.
DiscusiónEl SS es una enfermedad poco frecuente de etiología desconocida cuya importancia reside en su marcada expresividad clínica, porque se considera un marcador de enfermedades sistémicas9, asociándose a diversas neoplasias.
Aunque la fisiopatología es desconocida estaría en relación con la dishomeostasis del estrés y, sobre todo, del cortisol, se trata de un estrés desequilibrado o disestrés, donde se produce una quiebra en el equilibrio energético ante el hecho incuestionable de dar una respuesta adecuada a un aumento significativo del estrés. La secreción de cortisol prepara al organismo para mantener una respuesta más sostenida al estrés si el estímulo persiste, pero también se segrega dehidroepiandrosterona (DHEA) que actuaría contrarrestando los efectos adversos del cortisol. En el SS se produce un disbalance entre niveles séricos de cortisol y DHEA detectándose mayores niveles de cortisol sin que exista aumento paralelo de la DHEA. Evidencias clínicas y bioquímicas sugieren que las citocinas tienen un papel etiológico5,10,11.
La aparición del SS en un varón con múltiples recaídas2,12 con localización dermatológica no clásica13,14, pero sobre todo con alteraciones hematológicas, debe orientar el diagnóstico a un origen paraneoplásico y más concretamente hematológico que es el más frecuente.
Existen en la literatura casos descritos de SS asociado a SMD con lesiones extracutáneas que evolucionan de forma crónica y recurrente con desenlace fatal1. Cuando existe afectación pulmonar, los cuadros neumónicos no son siempre infecciosos, ya que, en raras ocasiones, algunos de ellos pueden ser inflamatorios. Los SS que se asocian a neoplasias suelen presentar previamente una infección respiratoria, las lesiones cutáneas son más graves y frecuentemente preceden al diagnóstico de la neoplasia; el 50% de los pacientes tienen lesiones extracutáneas y más de la mitad de estos no presentan neutrofilia, pero sí es frecuente encontrar anemia y anomalías plaquetarias8 con una recurrencia mayor del 70%. Se deben evaluar cuidadosamente los hemogramas, sobre todo cuando tienen afectación multilínea2. La anemia macrocítica con una evidencia morfológica de displasia, es muy sospechosa que pueda originarse por el SMD. El estudio microbiológico y anatomopatológico del lavado broncoalveolar son de gran utilidad en el diagnóstico de la alveolitis neutrofílica8 que es prácticamente una constante en el SS asociado a la mielodisplasia.
Las manifestaciones extracutáneas sugieren que este síndrome es un patrón de respuesta sistémica15–17. La cinética de los reactantes de fase aguda16,18 y su medición combinada aumentan la rentabilidad diagnóstica, alcanzando valores estadísticamente significativos que permiten conocer la evolución durante los brotes.
Así como los brotes de SS responden al tratamiento esteroideo, cuando se asocia a SMD tratado con dosis elevadas de corticoides y durante largo tiempo, el descenso12,19 de los mismos puede ser la causa de insuficiencia adrenal secundaria debido a la deprivación esteroidea y la consiguiente reactivación de la enfermedad, pudiendo ser este el origen de prácticamente la totalidad de los rebrotes sufridos por el paciente, que evolucionó de forma atípica, por su cronicidad y largo tiempo de enfermedad, con un desenlace fatal en relación con la presencia de síntomas sistémicos como parte de un proceso inflamatorio inespecífico que afectaba a diferentes órganos15. Aunque los corticoides sistémicos son el único tratamiento efectivo, en las formas paraneoplásicas los sucesivos rebrotes requieren dosis continuas y crecientes, que contribuyen al desarrollo del síndrome de Cushing iatrogénico.
Es fundamental mejorar el manejo clínico de estos pacientes para reducir el número de brotes del SS paraneoplásico y disminuir las recaídas por la mielodisplasia. Del mismo modo, habría que mejorar el uso de los corticoides para limitar los efectos secundarios propios del síndrome de Cushing, así como reducir las crisis addisonianas por la supresión brusca de los mismos u otras causas de desequilibrio esteroideo. Reducir también las descompensaciones diabética, hidroelectrolítica e infecciones por la inmunosupresión. Por último, el diagnóstico de aquellos enfermos afectos de un proceso menos evolucionado consistirá en la prevención de una evolución tórpida y que aporte calidad de vida a los pacientes afectos de un SS paraneoplásico.
