






Presentamos el caso de un paciente masculino con síndrome de Sjögren cuya manifestación clínica inicial fue extraglandular, teniendo el riñón como principal órgano afectado bajo la forma de acidosis tubular renal distal, que lo llevó a nefrocalcinosis como complicación en su evolución natural. Su hallazgo permitió el diagnóstico de esta exocrinopatía autoinmune, lográndose la remisión clínica y estabilización de la función renal con el uso de esteroides.
We describe a male patient with Sjögren's syndrome, whose initial clinical manifestation was extraglandular. The kidney was the main organ affected in the form of distal renal tubular acidosis that led to nephrocalcinosis as a complication during its natural progression. These findings led to the diagnosis of autoimmune exocrinopathy, with clinical remission and stabilization of renal function being achieved with the use of steroids.
El síndrome de Sjögren (SS) primario es la forma de presentación en el 50% de los pacientes, tiene una prevalencia aproximada de 0.5–3% en la población general, siendo la segunda enfermedad inflamatoria más frecuente después de la artritis reumatoide, sin contar con que un porcentaje alto de los casos pasa desapercibido; el SS es 9 veces más frecuente en mujeres que en hombres y tiene dos picos de presentación: el primero en la tercera década de la vida y el segundo durante la quinta, luego de la menopausia1,2. El SS secundario es más frecuente asociado a artritis reumatoide, entidad a la que se sobrepone entre el 20 y el 50% de los casos; también puede encontrarse asociado a lupus eritematoso sistémico, esclerosis sistémica progresiva, cirrosis biliar primaria y dermatomiositis3.
Aunque en 1888, Mickulicz describe un caso de un granjero con sequedad de mucosas, no fue sino hasta en 1930, cuando el oftalmólogo suizo Henrik Sjögren informó y englobó hallazgos clínicos y patológicos de forma sistémica al catalogarlos como compatibles con SS en 19 mujeres, 13 con diagnóstico probable de artritis reumatoide4.
Las manifestaciones clínicas, suelen ser variables, incluyendo manifestaciones extraglandulares como la afección renal, dentro de las que se describen la acidosis tubular renal distal (ATRD) y la nefrocalcinosis como complicación de esta, en su evolución natural.
La ATRD es un trastorno del equilibrio ácido-base definido por la incapacidad renal de compensar la generación de ácidos del metabolismo proteico, mediante su eliminación, en ausencia de un filtrado glomerular disminuido. El bicarbonato plasmático disminuye, aumenta el cloruro y la compensación respiratoria desciende la pCO25.
Existen pocos casos en publicaciones médicas de nefrocalcinosis en síndrome de Sjögren, siendo el nuestro del género masculino, en el cual, aunque se detectó tardíamente, su tratamiento con inmunosupresores permitió la reversibilidad de las manifestaciones clínicas, lográndose una estabilización de la función renal y lentificando su progresión a enfermedad renal crónica terminal.
Caso clínicoSe trata de un paciente masculino, de 33 años de edad, originario del Distrito Federal, con el antecedente patológico de 3 hermanos con diabetes mellitus tipo 2 y madre con hipertensión arterial sistémica. En 2008 fue referido a nuestro Instituto, procedente de un servicio de medicina interna de un hospital privado, con diagnóstico de síndrome de debilidad secundaria a hipopotasemia de etiología no determinada, sin embargo, el paciente no acudió a valoración, permaneció tomando sales de potasio de forma irregular durante los siguientes 2 años previos a acudir a nuestra urgencia, refiriendo episodios transitorios de debilidad recurrentes en promedio 10 por año e “infecciones urinarias” tratadas con antibióticos convencionales hasta 5 veces al año.
Acude al servicio de urgencias, con historia de 10 días de debilidad progresiva en extremidades, fatiga, astenia, mialgias con odinofagia y febrículas, negando disuria, nicturia, polaquiuria. Se automedica con sales de potasio sin mejoría, por lo que decide buscar ayuda médica. Al examen físico, se encontró con presión arterial de 119/66mmHg, frecuencia cardíaca de 67 latidos/minuto, frecuencia respiratoria de 22 ciclos/minuto, temperatura de 37°C, hiperemia faríngea y amigdalina, sin adenopatías ni otro hallazgo clínico anormal. Sus laboratorios al ingreso (tabla 1) reportaban una hipopotasemia leve, acidosis metabólica anión GAP normal, orina alcalina, sin glucosuria, con sedimento urinario con leucocituria, sin nitritos, ni bacterias evidentes a la microscopía, se envió a cultivar orina; nivel de complemento C3, C4 y CH50 normales, perfil viral negativo para VIH, virus hepatitis B y C. Durante la hospitalización, se profundizó en su estudio de extensión, entre los que destacó un ultrasonido renal con imágenes hiperecogénicas sugestivas de calcificaciones medulares (fig. 1), calciuria de 4.2mg/kg/día, anión GAP urinario positivo, fracción excretada de potasio de 12%, fracción excretada de bicarbonato 7%, pero un PH urinario que no logró acidificarse con una administración oral de cloruro de amonio, por lo que se catalogó como una acidosis tubular renal distal (tipo 1). Al reportarse urocultivo negativo, se decidió realizar biopsia renal percutánea, bajo la sospecha de una nefritis intersticial subyacente, basado en la proteinuria leve, leucocituria persistente, eritrocituria e hiperazoemia leve, la que reportó nefritis tubulointersticial crónica con lesiónlinfoepitelial tubular por células plasmáticas, nefrocalcinosis (intratubular e intersticial), fibrosis intersticial grado I y arterioloesclerosis leve (figs. 2 y 3), por lo que se inició tratamiento con prednisona 1mg/kg/día y citrato de potasio. Una semana después, existiendo reducción en leucocituria, se reportan estudios inmunológicos con anticuerpos antinucleares positivos, con un título de 1:2560, anti-SSA y anti-SSB positivos. Se cataloga, finalmente, como acidosis tubular renal distal secundaria a síndrome de Sjögren, se solicitó valoración a reumatología que coincide en el diagnóstico, agregando que se trata de un tipo primario o idiopático, se realizó test de Schirmer resultando positivo, curiosamente sin referir síntomas de ojo seco. En su seguimiento por consulta externa, por nefrología y reumatología, se ha logrado a las 16 semanas, reducir la dosis de esteroides hasta 5mg/día, con una recuperación del nivel de creatinina hasta 0.88mg/dL, reducción impresionante del requerimiento de sales de potasio manteniendo niveles de potasio normales, PH urinario alrededor de 5, calciuria de 3.9mg/kg/día, con ausencia de leucocituria, eritrocituria y proteinuria.
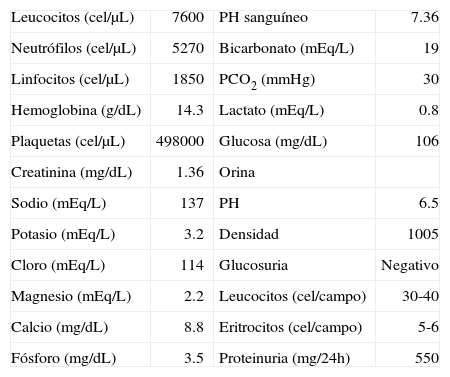
Laboratorios clínicos al ingreso.
| Leucocitos (cel/μL) | 7600 | PH sanguíneo | 7.36 |
| Neutrófilos (cel/μL) | 5270 | Bicarbonato (mEq/L) | 19 |
| Linfocitos (cel/μL) | 1850 | PCO2 (mmHg) | 30 |
| Hemoglobina (g/dL) | 14.3 | Lactato (mEq/L) | 0.8 |
| Plaquetas (cel/μL) | 498000 | Glucosa (mg/dL) | 106 |
| Creatinina (mg/dL) | 1.36 | Orina | |
| Sodio (mEq/L) | 137 | PH | 6.5 |
| Potasio (mEq/L) | 3.2 | Densidad | 1005 |
| Cloro (mEq/L) | 114 | Glucosuria | Negativo |
| Magnesio (mEq/L) | 2.2 | Leucocitos (cel/campo) | 30-40 |
| Calcio (mg/dL) | 8.8 | Eritrocitos (cel/campo) | 5-6 |
| Fósforo (mg/dL) | 3.5 | Proteinuria (mg/24h) | 550 |

 dando aspecto en “fila india” con atrofia tubular y pérdida del patrón “espalda con espalda” normal de los túbulos, a mayor aumento (B).")
Histopatología renal, en la que se muestra un infiltrado inflamatorio linfoplasmocitario intersticial con distribución en parches bajo la forma de agregado linfoide (A) dando aspecto en “fila india” con atrofia tubular y pérdida del patrón “espalda con espalda” normal de los túbulos, a mayor aumento (B).
; el estudio de inmunofluorescencia directa resultó negativo para todos los inmunoreactantes (B).")
El SS, constituye una exocrinopatía autoinmune muy rara en personas del género masculino, algunos describen incluso una razón hombre/mujer de 1:206,7. Con respecto a sus hallazgos clínicos, hasta un tercio de los pacientes pueden presentar manifestaciones extraglandulares, más activas y graves que las glandulares y que condicionan el pronóstico de la enfermedad a largo plazo8. La enfermedad extraglandular, se subdivide en no visceral (sistema músculo-esquelético) y en visceral (pulmón, corazón, tracto gastrointestinal, riñón, sistema nervioso, endocrino y hematológico). Nuestro paciente, prácticamente debutó con manifestaciones renales, aproximadamente durante 3 años, previas a diagnosticar la xeroftalmía, algo muy raro en esta enfermedad autoinmune.
A pesar que la xeroftalmía suele estar presente en 5–17% de la población adulta, es una manifestación prominente, muchos de los pacientes no están al tanto de los síntomas con los que se presenta, dentro de los que pueden mencionarse la sensación de cuerpo extraño, ojo rojo, fotofobia, fatiga ocular, dolor ocular, pestañas mal alineadas e intolerancia al uso de lentes de contacto9. Así mismo, el médico al evaluar el paciente debe determinar signos objetivos usando métodos para verificar la integridad de la superficie corneal y la producción de lágrimas10. Aunque la sensibilidad y la reproducibilidad del test de Schirmer son bastante bajas, es una forma objetiva y fácil de evaluar un paciente; por lo que algunos autores consideran que todo paciente con SS requiere una evaluación oftalmológica periódica1. En nuestro caso, su historial clínico no reportaba manifestaciones oculares, pero al evaluar su producción de lágrimas, curiosamente, se encontró con una reducción de ellas.
Las manifestaciones renales, aunque son descritas en la literatura en alrededor del 5% usualmente de forma silente, los estudios de series de casos reportan valores variables dependiendo de la búsqueda intencionada que se realice, así tenemos que los hallazgos catalogados como acidosis tubular renal, han sido encontrados entre 3.6% hasta 73.1% en las series de pacientes con SS estudiadas7,11, constituyendo la nefritis tubulointersticial aguda y/o crónica la forma histopatológica más común entre el 54 y el 80% de los casos11–14, seguida de las glomerulopatías en 45–50% de las series publicadas, predominando en orden de frecuencia la glomerulonefritis proliferativa focal, la glomeruloesclerosis focal segmentaria, la membranosa y membranoproliferativa13–15.
En la ATRD, o tipo 1, existe un defecto en la acidificación distal en las células alfa intercaladas del túbulo colector cortical (TCC); la eliminación urinaria neta de ácido (H+) se realiza en proporción de dos tercios mediante el amonio (NH4+) y un tercio mediante la acidez titulable (H2PO4–), los cuales son bajos, haciendo que el pH urinario esté anormalmente elevado para el grado de acidosis sistémica, en un contexto de una reabsorción de bicarbonato normal. En la acidosis se observa también pérdida renal de potasio y calcio secundariamente, hipocitraturia grave por reabsorción compensatoria y proteinuria de bajo peso molecular. La hipercalciuria multifactorial (liberación del calcio óseo, supresión del receptor sensor de calcio CasR, aumento de la carga distal de sodio y acidosis), junto con la orina alcalina e hipocitraturia favorecen la litiasis y nefrocalcinosis5,16. El filtrado glomerular, inicialmente normal puede disminuir evolutivamente por deshidratación, nefrocalcinosis, litiasis obstructiva y/o infección asociadas5,15.
La ATRD se acompaña de hipercloremia, como consecuencia de la disminución del bicarbonato en la sangre, debido al consumo por los ácidos circulantes; sin embargo, el riñón puede compensar esta pérdida gracias a que produce y reabsorbe el bicarbonato, por ello su excreción urinaria se mantiene baja (< 5% del filtrado) reflejo de que la absorción tubular de bicarbonato es normal a diferencia de la acidosis tubular proximal o tipo 217. En los túbulos proximales se reabsorbe la glutamina circulante a partir de la cual se sintetizan simultáneamente amonio y bicarbonato; por otro lado, la presencia de múltiples sistemas de transporte en la nefrona, hace posible recuperar alrededor del 95% del bicarbonato filtrado antes de llegar al túbulo distal, ello requiere un cotransportador Na+/HCO3– (NBCe1); esta absorción esta acoplada a la secreción de ácido en la orina por el intercambiador Na+/H+ (NHE3)17,18.
Las manifestaciones renales, son variables, dependiendo de la evolución de la enfermedad (aguda o crónica), la edad del paciente (hereditaria o adquirida), sus comorbilidades acompañantes (primaria o secundaria), sin embargo, su espectro incluye, de las formas leves con hipostenuria y cambios leves en el PH urinario, hasta cuadros graves presentes a las semanas de vida en las formas hereditarias, con poliuria, vómitos, episodios de deshidratación y retraso pondero-estatural con o sin sordera. En adultos, existen otras manifestaciones como hipopotasemia, debilidad muscular secundaria que puede progresar a parálisis flácida reversible, nefrocalcinosis, nefrolitiasis y osteomalacia/raquitismo al no recibir tratamiento5,16.
En el análisis de laboratorio, se encuentra, acidosis metabólica hiperclorémica con anión gap plasmático normal, normo o hipopotasemia, PH urinario anormalmente alcalino (> 5.5) en situación de acidosis, lo que refleja el defecto secretor distal de H+ secundario a la reducción en la formación de amonio y ácidos titulables, la cual constituye en forma de sales, la forma normal de eliminación neta de ácido en orina; por ello, el anión gap urinario resulta positivo16. En concordancia, con su etiología secundaria más común, como es el SS, la positividad de los anticuerpos antinucleares suele ser superior al 80%, asociándose con mayor frecuencia con afectación pulmonar y fenómeno de Raynaud. Los Ac anti-Ro/SS-A y anti-La/ SS-B se consideran los más específicos para el diagnóstico del SS aunque aparezcan en porcentajes variables (30–70%)19. Nuestro paciente no tenía manifestaciones de Raynaud, pulmonares, ni hallazgos de otra enfermedad autoinmune asociada, las cuales se buscaron intencionadamente.
Para los casos cuyo diagnóstico está en duda, como en la ATRD incompleta, término usado para describir la nefrocalcinosis o urolitiasis sin acidosis metabólica en estos pacientes, es recomendable aplicar pruebas de acidificación. Estas pruebas consisten en la administración de cloruro de amonio (NH4Cl), determinándose a continuación: el pH, la acidez titulable y la excreción urinaria de amonio20. La prueba a corto plazo, utilizada en gran parte de los estudios revisados, se administra 0,1g/kg de ClNH4 en dosis única y determinaciones horarias durante las ocho horas siguientes. En los casos en que se contraindique su uso (cirrosis hepática) podremos evaluar también la acidificación administrando cloruro cálcico (2mEq/kg)21. Nosotros documentamos a través de la prueba de acidificación, la incapacidad de la nefrona para reducir el PH urinario, estableciendo así un diagnóstico exacto del paciente.
Su tratamiento, debe llevar como meta normalizar la calciuria y la citraturia, para evitar la nefrocalcinosis y el daño renal progresivo, para ello, se debe corregir la acidosis subyacente a través de sales de bicarbonato o citrato de potasio y sodio, a una dosis suficiente para neutralizar toda la producción de ácido corporal (H+), que en adultos, usualmente requiere una dosis de 1mEq/kg/d fraccionada en 3–4 tomas/ día22. Sin embargo, este tratamiento constituye una medida paliativa cuando su causa es secundaria, como en el SS, donde se han usado corticosteroides y ciclofosfamida, según la severidad del involucro renal, con reportes de estabilización de la función renal, reducción de proteinuria y reversibilidad de varias manifestaciones clínicas derivadas de la acidosis tubular renal, evitando la extensión de la nefrocalcinosis y reduciendo el riesgo de progresión a estadio terminal de la enfermedad renal crónica11–13,23. En nuestro caso, se usó prednisona con pauta reductiva lenta, logrando una remisión de las manifestaciones clínicas al haber controlado la nefritis tubulointersticial autoinmune derivada del SS.
Esto debe ir seguido de un monitoreo adecuado y frecuente de parámetros de laboratorio hasta alcanzar un buen control terapéutico para reducir su riesgo de progresión a enfermedad renal crónica24.
ConclusiónLa ATRD es más común en la práctica clínica de lo que se cree, su detección temprana permite corregir los desequilibrios ácido-base y electrolíticos asociados, reduciendo el riesgo de complicaciones irreversibles como la nefrocalcinosis que indudablemente llevan a una enfermedad renal crónica en su evolución natural sin intervención terapéutica.
Los autores declaran no tener ningún conflicto de intereses.
