







Interstitial Lung Disease Associated with Autoimmune Diseases
Más datosInterstitial lung disease (ILD) is a common and serious manifestation of autoimmune rheumatic diseases. While the prevalence of ILD differs among the individual autoimmune rheumatic diseases, ILD remains an important cause of morbidity and mortality in systemic sclerosis, systemic lupus erythematosus, mixed connective tissue disease, primary Sjögren's disease, rheumatoid arthritis, and idiopathic inflammatory myositis. The present review summarizes recent literature on autoimmune-associated ILD with a focus on screening and monitoring for ILD progression. Reflecting on the currently available evidence, the authors propose a guideline for monitoring for progression in patients with newly diagnosed autoimmune-associated ILD. This review also highlights clinical and biological predictors of progressive pulmonary fibrosis and describes opportunity for further study in the rapidly evolving area of rheumatology and pulmonology.
La enfermedad pulmonar intersticial (EPI) es una manifestación común y seria de las enfermedades autoinmunes. Aunque la prevalencia de EPI difiere de acuerdo a cada enfermedad, continúa siendo una causa importante de morbilidad y mortalidad en la esclerosis sistémica, la artritis reumatoide, el síndrome de Sjögren, la enfermedad mixta del tejido conjuntivo y las miopatías inflamatorias. Este artículo de revisión resume la literatura reciente sobre la EPI asociada con autoinmunidad, con enfoque en la búsqueda y el monitoreo de la progresión de la EPI. Con base en la evidencia disponible, los autores proponen una guía para el monitoreo de la progresión en pacientes con la EPI asociada con autoinmunidad de reciente diagnóstico. Esta revisión también aborda los predictores clínicos y biológicos de la fibrosis pulmonar progresiva y resalta la oportunidad para estudios adicionales en áreas de rápida evolución como la reumatología y la neumología.
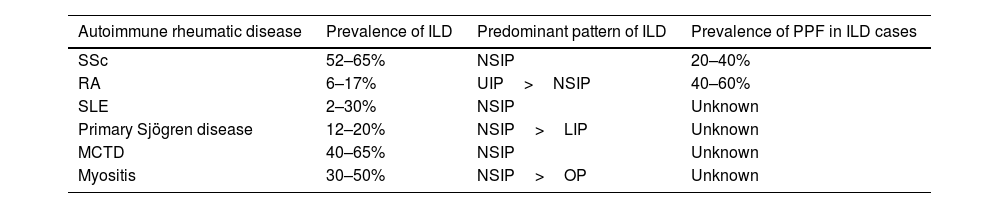
Pulmonary manifestations occur across the spectrum of systemic autoimmune rheumatic diseases and may affect the interstitium, vasculature, airways, and pleura. Interstitial lung diseases (ILD) represent an important cause of morbidity and mortality in patients with autoimmune rheumatic diseases.1 While the prevalence of ILD in individual autoimmune rheumatic diseases (e.g., systemic sclerosis [SSc], systemic lupus erythematosus [SLE], mixed connective tissue disease [MCTD]), primary Sjögren's disease [pSjD], rheumatoid arthritis [RA], and idiopathic inflammatory myositis [IIM]) varies2 (Table 1), studies have demonstrated that a substantial percentage of patients with autoimmune-associated ILD will develop progressive pulmonary fibrosis (PPF), a serious and often fatal condition.3
Prevalence and nature of ILD across the spectrum of autoimmune rheumatic diseases.
| Autoimmune rheumatic disease | Prevalence of ILD | Predominant pattern of ILD | Prevalence of PPF in ILD cases |
|---|---|---|---|
| SSc | 52–65% | NSIP | 20–40% |
| RA | 6–17% | UIP>NSIP | 40–60% |
| SLE | 2–30% | NSIP | Unknown |
| Primary Sjögren disease | 12–20% | NSIP>LIP | Unknown |
| MCTD | 40–65% | NSIP | Unknown |
| Myositis | 30–50% | NSIP>OP | Unknown |
Abbreviations: ILD=interstitial lung disease; SSc=systemic sclerosis; RA=rheumatoid arthritis; SLE=systemic lupus erythematosus; MCTD=mixed connective tissue disease; UIP=usual interstitial pneumonia; NSIP=nonspecific interstitial pneumonia; LIP=lymphocytic interstitial pneumonia; OP=organizing pneumonia.
The diverse pathological features of autoimmune-associated ILDs (e.g., fibrosis, inflammation, autoimmunity, and vasculopathy) engender immense clinical heterogeneity in both presentation and disease course. For example, some patients with autoimmune-associated ILD may have a stable course of disease, even in the absence of therapy, whereas other patients may experience PPF despite therapy. Recent advances in the understanding of ILD pathogenesis and distinct ILD clinical phenotypes, combined with an expansion of the ILD therapeutic pipeline, has made this field one of the most actively evolving fields in rheumatology.
The present narrative review summarizes recent research on autoimmune-associated ILD with a focus on screening, diagnosis, and monitoring for ILD progression. This review highlights the diverse clinical phenotypes of ILD within specific autoimmune rheumatic diseases and describes opportunities for further discovery. Articles were selected for inclusion in this review if they were published within the last 5 years. Select historical studies were included if they were deemed high quality and/or contained information not represented in the more recent literature cited.
Overview of diagnosis of autoimmune-associated ILDThe diagnostic evaluation of patients with autoimmune-associated ILD requires multi-disciplinary collaboration and a coordinated approached to decision making. A multi-disciplinary meeting (MDM) involving pulmonologists, rheumatologists, radiologists, and pathologists represents the gold standard for ILD diagnosis.4–6 The MDM provides a forum to discuss individual patient cases and review relevant clinical, radiological, laboratory and pathological findings. Because different radiological and histopathological patterns of ILD occur in patients with autoimmune rheumatic diseases (Fig. 1), expert input from radiologists and pathologists may help resolve diagnostic dilemmas. Additional members of an ILD–MDM may include psychologists, social workers, respiratory therapists, transplant specialists, thoracic surgeons, interventional pulmonologists, and palliative care providers. Studies have demonstrated improved diagnostic confidence and interobserver agreement after MDM compared with the individual components of an MDD (e.g., pathology, radiology).7–9 For the diagnosis of autoimmune-associated ILD, rheumatologists play an important role in guiding diagnostic decision making.10,11 For example, one study reported that approximately 10% of patients received a new diagnosis of an autoimmune-associated ILD after undergoing a MDM.7 In another study, rheumatology assessment reclassified 21% of patients with IPF as CTD-ILD.12
Approach to monitoring autoimmune ILD progression
Once a diagnosis of autoimmune-associated ILD is made, patients require vigilant monitoring for ILD progression. Different methods exist for monitoring for ILD progression including symptom and exercise tolerance evaluations, as well as physiological and radiological assessments. In 2023, an international clinical practice guideline defined PPF as an individual with ILD (other than idiopathic pulmonary fibrosis [IPF]) who experiences at least two of the following three criteria within the past year, not attributable to an alternate cause: (1) worsening respiratory symptoms; (2) physiological evidence of disease progression, defined as an absolute decline in FVC≥5% predicted and/or absolute decline in DLCO corrected for hemoglobin≥10%; (3) radiological evidence of disease progression.13
The guidelines do not specify the timing of symptom, physiological, and radiological assessments. Our opinion is that within the first 5 years of a new diagnosis of an autoimmune-associated ILD, patients should undergo close monitoring to understand the clinical phenotype of the ILD (e.g., stable, gradually versus rapidly progressive) (Fig. 2). While research efforts are underway to develop prediction tools for identifying those patients with an autoimmune-associated ILD who are most likely to develop PPF,14 this is an area of significant unmet need. Multiple modifiable and unmodifiable factors may affect ILD progression rates, including the underlying autoimmune rheumatic disease, its treatment, as well as co-morbid conditions. Understanding the nature of the ILD through close follow up during the first 5 years after a diagnosis may improve the personalization of treatment strategies and help the patient better understand their prognosis. Beyond 5 years, less vigilant monitoring may be considered if the patient has demonstrated ILD stability/improvement.
Systemic sclerosis
ILD occurs in the majority of patients with SSc.15 While the reported prevalence of ILD in SSc varies according to study design and patient population,16 one study demonstrated that among 1168 patients with SSc, 65% had evidence of ILD on HRCT.17 The most common histological and radiological pattern of ILD in SSc is non-specific interstitial pneumonia (NSIP), although usual interstitial pneumonia (UIP) and mixed patterns can also occur.18,19 The precise prevalence of PPF in SSc is not clear as previous studies have applied different definitions PPF. However, recent observational studies in largely European cohorts have demonstrated that the prevalence of ILD progression in SSc ranges from 27 to 38.5%.20 Among patients with SSc-ILD receiving treatment in a clinical trial in the US (Scleroderma Lung Study II, which compared mycophenolate versus cyclophosphamide),21 the rate of PPF was approximately 20%.14 Respiratory failure due to PPF is the leading cause of death in SSc.22
Because ILD occurs in most patients with SSc and is the leading cause of SSc-related death, universal screening for ILD is recommended (Table 2). HRCT of the chest is the preferred screening method, as a number of studies have demonstrated that pulmonary function tests (PFTs) may be normal early in the ILD course.23–25 In one prospective study of 102 patients with SSc, 62.5% of patients with ILD identified on HRCT had a normal FVC%-predicted.23
Proposed ILD screening approach in different autoimmune rheumatic diseases.
| Screen all patients with HRCT at time of diagnosis | Screen select patients with HRCT if risk factors for ILD present | Screen only those patients where there is a clinical suspicion for ILD |
|---|---|---|
| SSc | RAa | Primary Sjögren disease |
| Myositis | SLE | |
| MCTD |
Risk factors for ILD in RA include: male sex, ever smoker, age>60 years, high-positive RF and/or anti-CCP (>3× upper limit of normal), high disease activity, and other extra-articular RA manifestation.
Abbreviations: HRCT=high-resolution computed tomography; ILD=interstitial lung disease; SSc=systemic sclerosis; RA=rheumatoid arthritis; MCTD=mixed connective tissue disease.
ILD screening is recommended for all patients with SSc, regardless of the presence of respiratory symptoms. Respiratory symptom assessment is challenging in SSc as symptoms, such as dyspnea and cough, can have multiple contributing etiologies (i.e., reflux disease causing cough). Moreover, a recent study of SSc-ILD patients assigned to the placebo arm of the nintedanib (SENSCIS) trial found that the rate of decline of FVC over 52 weeks was similar irrespective of the presence of cough or dyspnea at baseline.26
Once diagnosed with ILD, all patients should be monitored closely for disease progression.27,28 Because distinct clinical phenotypes of ILD progression exist in SSc,29 the monitoring approach may vary based on the individual patient. For example, the presence of certain factors (e.g., male sex, anti-Scl-70 antibody positivity) increases the risk of SSc-PPF.30 However, research on risk stratification in SSc-ILD is still evolving. For example, while historical studies demonstrated that the greatest decline in lung function occurred early in the ILD course (with the first 2 years),31 more recent studies have illustrated that ILD also progresses at later stages in the SSc disease course.32
Emerging research from both observational and clinical trial studies has identified new risk factors for ILD progression in SSc. These factors include increased severity of reflux disease,32,33 as well as elevated circulating levels of the pneumoproteins Krebs von den Lungen 6 (KL-6) and chemokine ligand 18 (CCL-18).34–36 The integration of clinical and biological data in risk stratification will undoubtedly improve our ability to predict SSc-ILD phenotypes at the time of diagnosis and tailor the ILD monitoring approach accordingly.
Systemic lupus erythematosusDiverse pulmonary manifestations may occur in patients with SLE, including pleuritis, pulmonary thromboembolism, diffuse alveolar hemorrhage, lupus pneumonitis, shrinking lung syndrome, as well as ILD. Estimates of ILD prevalence in SLE vary widely, with some studies demonstrating a prevalence of only 2–4% to almost 30%.37 A recent meta-analysis reported a prevalence range of 3–10%.2 A nationwide population-based study conducted in France found that among SLE patients admitted to the hospital between 2011 and 2012, 1.2% had a diagnosis of ILD.38 In those patients with SLE who did not have ILD at baseline, ILD subsequently occurred in 2.6% of patients between 2013 and 2020.38
Risk factors for ILD in patients with SLE include older age39 and longer disease duration.40 In a study of over 3000 patients with SLE, the mean disease duration of SLE at the time of ILD diagnosis was 7.7 years.40 Overall, 22% of patients with a history of acute lupus pneumonitis and 21% of patients with a history of alveolar hemorrhage who survived subsequently developed ILD.40
The most common radiographic pattern of ILD in SLE is NSIP, accounting for 40–55%.39,41,42 Other radiographic patterns may include UIP, organizing pneumonia (OP), as well as lymphocytic interstitial pneumonia (LIP). In terms of histopathology, one small study demonstrated that an unclassifiable pattern (defined as NSIP and OP) was the most common pattern observed in 12 patients with SLE who underwent surgical lung biopsy or autopsy.41
The prevalence of PPF in patients with SLE is unclear. Longitudinal cohort studies of patients with SLE-ILD are scarce. One study of 20 patients with SLE-ILD revealed that radiological progression occurred slowly, and physiological measures remained stable over varying follow up periods.39 Another study of 55 patients with SLE-ILD demonstrated a 5-year survival of 85.3%.41 After adjusting for age, other risk factors for mortality in this cohort included current smoker, elevated serum KL-6, increased extent of ILD on HRCT, thrombocytopenia, anti-dsDNA antibody titer, and thrombocytopenia.41 In the aforementioned population-based study in France, ILD presence was associated with an increased risk of death in the multivariable analysis.38
Universal screening for ILD with HRCT of the chest in patients with SLE is currently not recommended. Future studies are needed to determine whether patients with SLE who possess certain features of SLE should undergo screening. Monitoring for ILD progression (Fig. 2) is paramount to ensuring that patients with SLE receive timely access to treatments for their ILD.
Mixed connective tissue diseaseMixed connective tissue disease (MCTD) is a condition characterized by high titer anti-U1-RNP antibodies in combination with overlapping clinical features of SSc, SLE and polymyositis/dermatomyositis.43,44 The diagnosis of MCTD is challenging due to the heterogeneity of the disease and similarities to other autoimmune disease.45 In recent studies, the prevalence of ILD in patients with MCTD ranges considerably from 9.1% to 56%.43 In a subset of patients with MCTD with predominant SSc features, ILD can occur in up to 61% of patients.46
The most common pattern of ILD seen in MCTD is NSIP,47 though UIP and organizing pneumonia can occur.48 In addition to predominant SSc features, male sex, elevated anti-RNP antibody titer, anti-Ro52, and anti-Smith are associated with an increased risk of ILD, whereas arthritis is associated with a lower ILD risk.49,50 Anti-RNP antibody titers correlate with increased disease activity in both adult and juvenile patients with MCTD.49,51 Recent studies suggest the presence of esophageal dysmotility, Raynaud phenomenon and myositis symptoms are each associated with an increased risk of ILD.50,52
Historically, the course of ILD in MCTD was deemed milder than other autoimmune ILDs, with modest changes in PFTs and HRCT findings reported over time.53 However, in a more recent, longitudinal observational cohort study in Norway, even mild changes in HRCT findings were associated with decreased lung function.49 The reported five-year survival for MCTD-ILD is estimated at 84.7%.1 ILD is associated with an increased risk of mortality in MCTD, and the radiological extent of ILD likely moderates this relationship. For example, patients with >10% total lung volume involvement on HRCT have a 10-year cumulative survival rate of only 60%.49
HRCT remains the gold standard for evaluation of ILD in MCTD54 and can be effective in monitoring disease response to therapy and progression.53–55 While no specific guidelines exist for screening and monitoring for ILD in patients with MCTD, given the higher prevalence of ILD in patients with features of SSc, one approach is to follow the most recent consensus guidelines on screening for SSc,28 which includes cardiopulmonary examination, spirometry with DLCO, HRCT of the chest and antibody testing at time of diagnosis. This approach has been proposed elsewhere.56
Primary Sjögren diseasePrimary Sjögren disease (pSjD) is an autoimmune disease typified by dysfunction of salivary and lacrimal glands resulting in sicca symptoms.57 Extraglandular manifestations of pSjD are common and may affect multiple organs including the lungs.58,59 Secondary Sjögren disease refers to the symptoms of Sjögren disease occurring in conjunction with another autoimmune disease, such as SSc, MCTD, and most commonly SLE.60 ILD occurs in patients with both pSjD and secondary Sjögren disease.
Recent estimates of the prevalence of ILD in pSjD vary considerably, ranging from 2.8 to 78.6%.1,61–64 NSIP is the most frequent pattern of ILD found in pSjD, but UIP, organizing pneumonia, LIP and, rarely, desquamative interstitial pneumonitis can also occur.65–67
Studies have demonstrated that older age, male sex, and elevated CRP are associated with higher risk of developing ILD in pSjd.64,67–70 Potential biomarkers of ILD in pSjD include serum levels of IgM, exotaxin, KL-6, TGFα, TNF,70 serum IgG levels,67 and elevated serum levels of tumor markers, most notably CA153.71 However, limited studies exist on biomarkers in pSjD-ILD, and some studies report contradictory evidence. Development of ILD in pSjD is associated with a 5-year survival of 84.7%1 and a 10-year survival of 81.7%.65
Clinical studies assessing the timing and modality for screening for ILD in pSjD are scarce. One study assessing the role of HRCT in pSjD patients without pulmonary symptoms found that 65% (N=24/37) of asymptomatic patients had abnormal HRCT findings.72 The most common abnormalities observed in this relatively small cohort included interlobular septal thickening, micronodules and ground glass attenuation. This same study found that the 7 of the 24 patients with abnormal HRCT findings had normal PFTs.72 Although there are no guidelines for screening for ILD in pSjD, one expert consensus statement on autoimmune-associated ILD recommended screening with PFTs if a patient with pSjD presents with respiratory symptoms.73 However, sicca symptoms themselves can trigger cough and the sensation of breathlessness, making clinical evaluation for lung disease challenging.74,75
Alternative screening approaches under consideration for pSjD include lung ultrasound (LUS). One small study (N=13) demonstrated that LUS can predict the presence of HRCT-defined ILD with good accuracy.76 While LUS may be feasible in a clinic-based setting and does not expose patients to excess radiation, it is highly operator dependent and requires further study.76
The prevalence of ILD in secondary Sjögren syndrome is unknown. In patients with SSc and secondary Sjögren syndrome, the ILD is often attributed to the underlying SSc, with rare exception (e.g., patient presenting with an LIP pattern on HRCT imaging). Interestingly, in other autoimmune diseases, such as SSc and myositis, the presence of the anti-SSA 52kDa IgG (also known as anti-Ro52) is associated with an increased risk of ILD.77,78
Among the limited number of longitudinal studies examining progression of pSjD-ILD, one recent retrospective study in South Korea (N=62) found that 50% of pSjD-ILD patients experienced disease progression over a 17-year observation period.79 Two additional longitudinal studies demonstrated progression of pSjD-ILD in 38.6% of patients (N=83) over a 5-year period66 and 20.4% of patients (N=49) over a 3-year period.67 It is presently unclear whether the presence of an UIP pattern of ILD is associated with progression of pSjD-ILD.66,67
Rheumatoid arthritisPulmonary manifestations of RA include ILD, bronchiectasis, nodules, pleuritis, and airway involvement.80 The relative risk of respiratory mortality is markedly increased compared to the general population,81–86 particularly for seropositive RA that has elevated rheumatoid factor (RF) and/or anti-cyclic citrullinated peptide (anti-CCP). Studies in people with elevated RF and/or anti-CCP have shown more lung damage and elevated autoantibodies in sputa that suggest pulmonary mucosa as a possible origin of RA-related autoantibodies.87,88
Unlike other autoimmune rheumatic diseases, RA seems to have a predilection for the UIP pattern of ILD,2 with over half of patients with RA-ILD having UIP. The UIP subtype is more likely to be progressive in patients with RA, compared to other inflammatory subtypes such as NSIP and OP.89 The MUC5B promoter variant is a common genetic polymorphism (10–15% of those with European ancestry) initially found to be associated with IPF.90 This is the strongest genetic variant associated with RA-ILD, particularly for the UIP subtype.91,92 Other potential genetic associations with RA-ILD include RPA3-UMAD1 (identified in a Japanese genome-wide association study),93 other IPF genetic risk variants (TOLLIP, IVD),91 telomere maintenance genes (TERT, PARN, RETL1),94 and surfactant homeostasis (SFTPC).94
The prevalence of RA-ILD varies widely based on study methodology. For example, research protocols that screen all patients for RA-ILD have found 15–40% of patients have imaging features of ILD.95,96 Retrospective studies that use clinically indicated chest CT may also find high prevalence of ILD features.80 A meta-analysis reported a pooled prevalence of 11% (95%CI 7–15%).2 A population-based study in Olmsted County, Minnesota estimated that the cumulative incidence of RA-ILD was 15.3% after 20 years of RA disease duration.97 Several studies suggest rising RA-ILD prevalence,98,99 perhaps due to increased surveillance/awareness. A study performed among smokers showed that subclinical ILD was present in 17% of RA versus 5% of non-RA comparators.100 Having RA and subclinical ILD was associated with 3-fold increased mortality.100 Some patients may present with ILD and later be diagnosed with RA, suggesting a bidirectional relationship of RA and ILD.101
There are no accepted guidelines on who should be screened for RA-ILD, but many potential risk factors exist, including male sex, older age (>60 years), older age at RA onset, elevated RF and/or anti-CCP (especially>3× upper limit of normal), and high articular disease activity.102–104 Some others may include obesity, poor functional status, and education.103 The literature on whether glucocorticoids and specific disease-modifying antirheumatic drugs (DMARDs) affect RA-ILD risk is controversial. For example, while methotrexate can rarely cause a drug-induced pneumonitis, most studies have not found any association between methotrexate exposure and increased risk of future ILD in RA.103,105–107 Presence of the MUC5B promoter variant is strongly associated with RA-ILD. This is one of the few risk factors specific to the UIP subtype.91 Other potential biomarkers108 include inflammatory and fibrotic markers such as KL-6, SP-D, MMP-7, PARC, Ip-10/CXCL10, and HSP90109–111 and autoantibodies such as fine specificity anti-citrullinated protein antibodies,112 anti-malondialdehyde–acetaldehyde,113 anti-peptidylarginine deiminase 3X4R,114 and anti-carbamylated protein antibodies.115 Research is ongoing to identify risk factors for specific RA-ILD subtypes, understand different natural history patterns on longitudinal follow-up, and determine the clinical utility of screening for ILD.116
Median survival after clinical RA-ILD onset is generally only 3–8 years,97,98,117 emphasizing the need for close monitoring and treatment. A recent meta-analysis reported that mortality within 5–10 years of RA-ILD diagnosis was 49%.118 There are no specific recommendations for monitoring RA-ILD, so most clinicians often use a similar approach as for IPF. Once diagnosed, most will require frequent pulmonary function tests (generally every 3–6 months) and at least annual HRCT.
Idiopathic inflammatory myositisIdiopathic inflammatory myositis (IIM) is comprised of heterogeneous disorders that generally affect the muscles, skin, and/or lungs.119 Dermatomyositis and anti-synthetase syndrome have a predilection for lung involvement.120 Some patients with IIM present with fulminant lung disease, including diffuse alveolar hemorrhage that has high short-term mortality.121 Some patients may also have respiratory failure due to respiratory and bulbar muscle weakness.122 Thus, pulmonary involvement in IIM is a major contributor to morbidity and mortality.123 Patients with IIM are generally screened for ILD and cancer.
In a recent systematic review and meta-analysis, the pooled prevalence of ILD within IIM was 41% (95%CI 33–50%).2 The most common subtype of ILD is NSIP, followed by OP, diffuse alveolar damage, and UIP.2 However, the prevalence, subtype, and severity vary markedly based on IIM subtype. For example, >90% of patients with anti-MDA5 dermatomyositis have lung involvement, most of which are progressive and have high mortality.124 Anti-synthetase syndrome with elevated anti-Jo1 also has high likelihood of ILD (>90%).125 Other clinical biomarkers that are associated with ILD in IIM include anti-PL7/12, anti-NXP2, anti-Tiff, Ro/SS-A (specifically Ro52), anti-KS, and anti-OJ.2,126 Myositis antibody panels that test for many autoantibodies are useful to risk stratify suspected IIM patients for ILD and cancer risk, while also identifying patients with anti-synthetase syndrome.126–128 The presence of clinical features such as mechanic's hands, amyopathic presentation, and the absence of malignancy also increase risk for ILD within IIM.2 In contrast, polymyositis, inclusion body myositis, and immune-mediated necrotizing myopathy generally have lower risk for ILD.2
Most longitudinal ILD studies in IIM have included patients with anti-Jo1 anti-synthetase syndrome and anti-MDA5 dermatomyositis and focused on treatment since these patients have an aggressive course.125,129,130 There are no current specific recommendations for monitoring ILD in IIM, but these patients typically need very frequent monitoring and low threshold for aggressive treatment due to the prevalence and severity of lung disease. The type of monitoring needed for patients without baseline abnormalities on either pulmonary function tests or HRCT is currently unclear. This likely may vary based on underlying risk factors. For example, IIM patients with elevated anti-MDA5 or anti-Jo1 antibodies should likely still need close monitoring with serial PFTs and/or chest CT.
Future directions and conclusionsConsidering the high prevalence of ILD in patients with autoimmune rheumatic diseases and its overall impact on quality of life and survival, there is an unmet need for improving screening, monitoring, and management. However, there are clear challenges related to the underlying heterogeneity and rarity within each disease, which make clinical trials and observational studies difficult to conduct. Also, more natural history studies are needed to define subtypes and trajectories of ILD within distinct autoimmune diseases. Biomarkers may be particularly helpful to identify people at risk for ILD progression, while MDM should be incorporated more broadly, including community settings given the prevalence and severity of autoimmune-associated ILD. Standardized screening questionnaires should be developed to capture symptoms in patients with early ILD, while also accounting for confounders such as obesity, deconditioning, and frailty, particularly in populations where joint damage may limit mobility. Finally, trials are needed to investigate how currently available DMARDs affect risk and progression of autoimmune-associated ILD. New ILD-targeted therapies are needed with improved efficacy and better tolerability. There are now several pharmacologic trials showing efficacy for ILD within specific patient populations, but important treatment gaps remain.
Authors’ contributionsSDG, JAS, ERV contributed equally to this article. Each author participated in conducting the literature search for this narrative review, writing the original draft of this manuscript and revising the manuscript. All authors approved the final manuscript prior to submission.
Conflict of interestThe authors report no financial or personal relationships related to the submitted work. Outside of the submitted work, the authors report the following relationships: SDG reports no declarations of interest; JAS is supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases (grant numbers R01 AR080659, R01 AR077607, P30 AR070253, and P30 AR072577), the R. Bruce and Joan M. Mickey Research Scholar Fund, and the Llura Gund Award for Rheumatoid Arthritis Research and Care. JAS has received research support from Bristol Myers Squibb and performed consultancy for AbbVie, Amgen, Boehringer Ingelheim, Bristol Myers Squibb, Gilead, Inova Diagnostics, Janssen, Optum, and Pfizer unrelated to this work; ERV is supported by the National Heart, Lung, and Blood Institute (grant number K23 HL150237) and reports the following financial relationships outside of the submitted work on autoimmune-associated ILD: Consulting (Boehringer Ingelheim, Roche, CSL Behring, GSK); Speaking (Boehringer Ingelheim); Institutional support received for performing studies in systemic sclerosis for Kadmon, Forbius, Boehringer Ingelheim, Horizon, Prometheus.
The following are the supplementary material to this article:
