











Las adenopatías generalizadas son una manifestación frecuente, aunque usualmente inespecífica e indicativa de múltiples enfermedades. Las enfermedades autoinmunes, entre ellas el lupus eritematoso sistémico, pueden tener como primera manifestación dicho hallazgo; por lo tanto se deben tener en cuenta para el diagnóstico diferencial. En los casos que se presentan, las adenopatías diseminadas encontradas clínica y radiológicamente, fueron una variable de confusión para el diagnóstico rápido y al que se logró llegar mediante el adecuado contexto clínico, paraclínico y el estudio histopatológico. Dado que el lupus es una enfermedad potencialmente fatal, es importante reconocer esta forma de presentación.
Generalised lymphadenopathy is a common manifestation, but usually non-specific and indicative of multiple diseases. Autoimmune diseases, including lupus erythematosus, may be the first manifestation of this finding, and therefore should be considered for differential diagnosis. In the cases presented, clinically and radiologically disseminated lymphadenopathy, they were confounding variables for rapid diagnosis, which is reached through the proper clinical and para-clinical context and with histopathological confirmation. Given that lupus is a potentially fatal disease, it is important to recognise this presentation.
Los nódulos linfáticos son un conglomerado de células que hacen parte del sistema de defensa; de los cuales se reconocen más de 6001, al presentar una anormalidad en el tamaño o en el carácter de los nódulos se denominan adenopatías2. Las adenopatías pueden obedecer a múltiples condiciones que se recuerdan fácilmente con el acrónimo «MIAMI» (Malignidad, Infecciones, Autoinmunidad, Misceláneas, Iatrogénicas)2; esta manifestación clínica puede configurar un reto diagnóstico para el clínico ya que, en ocasiones, el amplio diferencial dificulta una aproximación etiológica clara y temprana.
El lupus eritematoso sistémico (LES) es una enfermedad autoinmune, potencialmente mortal, que en muchas ocasiones es confundida con otras condiciones; se estima una prevalencia aproximada de 40 casos por cada 100.000 personas3. La frecuencia de aparición de adenopatías en LES es variable, reportándose un rango de 25 a 50%4, estas se asocian con diferentes características clínicas y paraclínicas de actividad de la enfermedad como eritema malar, úlceras orales, esplenomegalia, alopecia, positividad de anti-DNA doble cadena, entre otras5.
Se presentan, a continuación, dos casos clínicos de pacientes que consultaron con adenopatías generalizadas como síntoma cardinal, en los cuales por la forma de presentación el primer diagnóstico diferencial fueron las enfermedades linfoproliferativas; sin embargo, un interrogatorio exhaustivo y un enfoque clínico racional llevaron al diagnóstico certero de LES, permitiendo un adecuado tratamiento.
Presentación de los casosCaso 1Paciente femenino de 23 años de edad procedente de Ibagué, quien consulta en marzo de 2012 por cuadro clínico de 3 meses de evolución, consistente en pérdida de peso de aproximadamente 20 kilos, asociado a sudoración nocturna, astenia, adinamia, dolores articulares de predominio en manos y hombros, acompañados de pérdida de la fuerza muscular en miembros inferiores, alopecia y fiebre de predominio nocturno.
Presenta como antecedentes médicos hipotiroidismo en suplencia. Al examen físico se encuentran adenopatías axilares por lo cual inician estudios de extensión con sospecha de síndrome linfoproliferativo versus tuberculosis ganglionar. Se toma biopsia de ganglio axilar izquierdo con informe de patología que reporta enfermedad linfoproliferativa sugestiva de proceso reactivo, caracterizada por folículos de diferentes tamaños fusionados, preservación del manto y centros germinales activos.
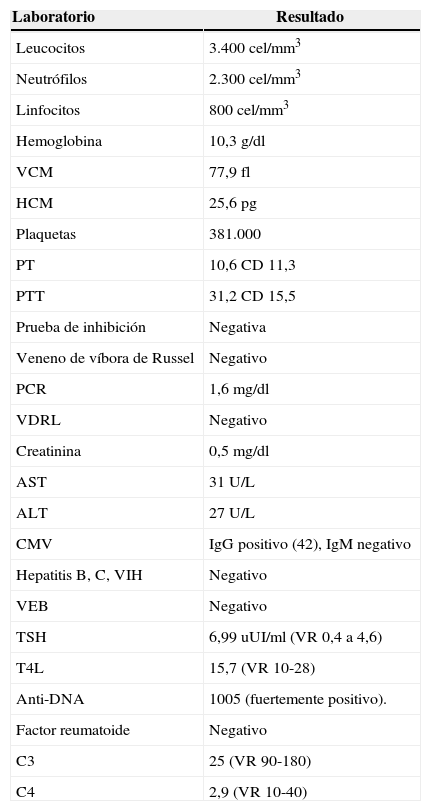
Con estos hallazgos la paciente consulta a nuestra institución en donde se hospitaliza y se inician estudios de extensión. Al examen físico llama la atención la presencia de adenopatías cervicales derechas, adenopatías inguinales bilaterales, abdomen con hepatomegalia. El reporte de la tomografía axial computarizada (TAC) de cuello informa múltiples ganglios subcentrimétricos bilaterales, el TAC de tórax describe adenomegalias axilares y el TAC abdominopélvico indica adenomegalias retroperitoneales (figs. 1–3). Se ordenan paraclínicos para descartar procesos infecciosos (tabla 1).



Reporte de laboratorio del paciente del caso 1
| Laboratorio | Resultado |
|---|---|
| Leucocitos | 3.400 cel/mm3 |
| Neutrófilos | 2.300 cel/mm3 |
| Linfocitos | 800 cel/mm3 |
| Hemoglobina | 10,3g/dl |
| VCM | 77,9 fl |
| HCM | 25,6pg |
| Plaquetas | 381.000 |
| PT | 10,6 CD 11,3 |
| PTT | 31,2 CD 15,5 |
| Prueba de inhibición | Negativa |
| Veneno de víbora de Russel | Negativo |
| PCR | 1,6mg/dl |
| VDRL | Negativo |
| Creatinina | 0,5mg/dl |
| AST | 31 U/L |
| ALT | 27 U/L |
| CMV | IgG positivo (42), IgM negativo |
| Hepatitis B, C, VIH | Negativo |
| VEB | Negativo |
| TSH | 6,99 uUI/ml (VR 0,4 a 4,6) |
| T4L | 15,7 (VR 10-28) |
| Anti-DNA | 1005 (fuertemente positivo). |
| Factor reumatoide | Negativo |
| C3 | 25 (VR 90-180) |
| C4 | 2,9 (VR 10-40) |
ALT: alanina aminotransferasa; AST: aspartato aminotransferasa; CMV: citomegalovirus; C3: componente 3 de complemento; C4: componente 4 del complemento; DNA: ácido desoxirribonucleico; HCM: hemoglobina corpuscular media; PCR: proteínaC reactiva; PT: tiempo de protrombina; PTT: tiempo parcial de tromboplastina; TSH: hormona estimulante de la tiroides; T4L: tiroxina libre; VCM: volumen corpuscular medio; VDRL: venereal disease research laboratory; VEB: virus de Epstein-Barr; VIH: virus de la inmunodeficiencia humana.
Se indica toma de biopsia excisional más estudio de inmunohistoquímica de ganglio axilar izquierdo, que muestra hallazgos morfológicos y de inmunofenotipo que corresponden a hiperplasia folicular reactiva florida; no hay evidencia de compromiso neoplásico ni formación de granulomas.
Con estos resultados se revalúa a la paciente considerando síndrome constitucional, síntomas B asociados a artralgias de más de 3 grupos articulares, alopecia, fenómeno de Raynaud (detectado en el nuevo interrogatorio); hemograma con bicitopenia (leucopenia y anemia microcítica hipocrómica heterogénea) y anti-DNA de doble cadena fuertemente positivo. Por lo anterior, se hace diagnóstico de LES, se inicia desparasitación, metilprednisona en bolos e hidroxicloroquina.
Caso 2Paciente femenino de 15 años de edad proveniente de Bogotá, estudiante de bachillerato, quien consulta por cuadro clínico de 2 meses de evolución, consistente en osteomialgias generalizadas, dorsolumbalgia, dolores articulares de predomino en manos, rodillas y pies, con posterior limitación para la marcha por dolor y estado de postración secundario; además presenta pérdida de peso de 10 kilos en los últimos 4 meses, caída del cabello y fiebre de 15 días de evolución.
Había estado hospitalizada extrainstitucionalmente, con hallazgos clínicos de adenopatías generalizadas asociadas a leucocitosis y deciden realizar biopsia de médula ósea la cual se encuentra dentro de los límites normales.
La paciente ingresa en el hospital por exacerbación de la sintomatología descrita y al examen físico se encuentra alerta, orientada, taquicárdica, afebril, normotensa, con palidez mucocutánea generalizada, adenopatías preauriculares derechas en conglomerado y cervicales móviles no dolorosas, fuerza muscular disminuida en miembros inferiores con hipotrofia de las extremidades.
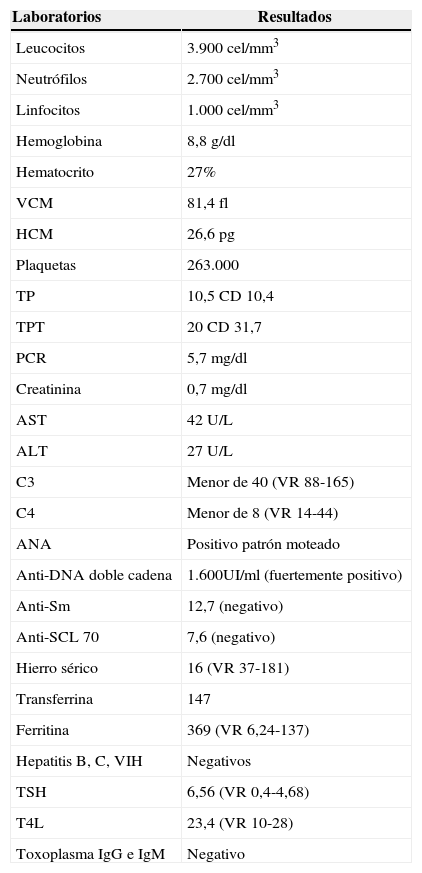
Con los hallazgos clínicos descritos se sospecha LES y se inician estudios de extensión (tabla 2). Se ordena TAC de cuello que informa adenomegalias bilaterales de predominio derecho, TAC de tórax que reporta pérdida de volumen y tracto fibroatelectásico locales con derrame pleural laminar y adenomegalias axilares bilaterales y TAC de abdomen que describe ganglios paraaórticos (figs. 4–7).
Reporte de laboratorio del paciente del caso 2
| Laboratorios | Resultados |
|---|---|
| Leucocitos | 3.900 cel/mm3 |
| Neutrófilos | 2.700 cel/mm3 |
| Linfocitos | 1.000 cel/mm3 |
| Hemoglobina | 8,8g/dl |
| Hematocrito | 27% |
| VCM | 81,4 fl |
| HCM | 26,6pg |
| Plaquetas | 263.000 |
| TP | 10,5 CD 10,4 |
| TPT | 20 CD 31,7 |
| PCR | 5,7mg/dl |
| Creatinina | 0,7mg/dl |
| AST | 42 U/L |
| ALT | 27 U/L |
| C3 | Menor de 40 (VR 88-165) |
| C4 | Menor de 8 (VR 14-44) |
| ANA | Positivo patrón moteado |
| Anti-DNA doble cadena | 1.600UI/ml (fuertemente positivo) |
| Anti-Sm | 12,7 (negativo) |
| Anti-SCL 70 | 7,6 (negativo) |
| Hierro sérico | 16 (VR 37-181) |
| Transferrina | 147 |
| Ferritina | 369 (VR 6,24-137) |
| Hepatitis B, C, VIH | Negativos |
| TSH | 6,56 (VR 0,4-4,68) |
| T4L | 23,4 (VR 10-28) |
| Toxoplasma IgG e IgM | Negativo |
ALT: alanina aminotransferasa; ANA: anticuerpos antinucleares; Anti-SCL 70: anticuerpos anti-topoisomerasa 1; Anti-Sm: anticuerpos anti-Smith; AST: aspartato aminotransferasa; C3: componente 3 de complemento; C4: componente 4 del complemento; DNA: ácido desoxirribonucleico; HCM: hemoglobina corpuscular media; PCR: proteínaC reactiva; PPT: tiempo parcial de tromboplastina; TP: tiempo de protrombina; TSH: hormona estimulante de la tiroides; T4L: tiroxina libre; VCM: volumen corpuscular medio; VIH: virus de la inmunodeficiencia humana.




Con estos hallazgos se hace diagnóstico de LES y se decide iniciar manejo con pulsos de metilprednisolona, presentando al tercer día mejoría significativa del estado clínico.
DiscusiónEn el presente estudio se realiza el reporte de dos casos clínicos de pacientes con linfadenopatías generalizadas que, por su forma de presentación, indujeron al diagnóstico inicial de neoplasia tipo linfoproliferativa, sin embargo, los hallazgos histológicos y la evaluación concienzuda de la anamnesis condujeron al diagnóstico acertado de LES, con la consecuente mejoría una vez se inició el manejo dirigido.
El diagnóstico diferencial de la enfermedad hematológica neoplásica, en estos pacientes, es lógico dados los síntomas constitucionales presentes y reconociendo que hasta el 80% de los linfomas tienen adenopatías en el curso de la enfermedad6. De manera adicional, se conoce el riesgo de concomitancia de las dos condiciones; reportándose en la literatura asociación con LES y otras condiciones autoinmunes como tiroiditis, síndrome de Sjögren, entre otros7. Landgren et al., en un estudio escandinavo de casos y controles concluye una fuerte correspondencia de LES y linfoma de Hodgkin con un odds ratio de 5,8 (IC de 2,2 a 15,1)8, así como mayor frecuencia de enfermedad de Castleman en este grupo de pacientes9. Apora et al., en un metaanálisis de cohortes prospectivas describe un aumento del riesgo de linfoma de Hodgkin, linfoma no Hodgkin, leucemia y mieloma cuando se comparó con la población general10.
Las linfadenopatías persistentes en pacientes con LES que no responden a tratamiento podrían ser indicativos de linfoma, como se comentó previamente. Varios estudios desde 1970 han reportado un riesgo incrementado de malignidad en enfermedades autoinmunes, este riesgo es secundario a la combinación de mutaciones en líneas germinales y somáticas, sobreestimulación inmune persistente e inmunosupresión farmacológica11,12.
Estas razones apoyan el hecho de que la biopsia de las adenopatías desempeñan un papel importante cuando la sospecha clínica lo amerita o cuando el paciente tiene un diagnóstico previo de LES y estas no mejoran a pesar del manejo adecuado. La asociación entre la actividad de la enfermedad autoinmune y el incremento del riesgo de estas neoplasias es contradictorio, dados los hallazgos de diferentes estudios observacionales13.
Entre las enfermedades autoinmunes, el LES es una enfermedad relevante ya que es potencialmente fatal y puede ser fácilmente confundida con muchos otros desórdenes. La prevalencia del lupus es de aproximadamente 40 casos por 100.000 personas en el norte de Europa y en Estados Unidos el número de pacientes con lupus excede los 250.0003. Las adenopatías asociadas a LES, en la mayoría de los casos, son localizadas y en pocas ocasiones se presentan como adenopatías generalizadas en el comienzo de la enfermedad, además en la validación de los últimos criterios de clasificación se excluyen como elemento diagnóstico14, con estos dos casos reportados evidenciamos que esta forma de presentación puede darse y que es importante considerarlo en el diagnóstico diferencial.
La edad está relacionada con la forma de presentación del LES y las adenopatías no escapan a esta característica, siendo más frecuentes cuando la enfermedad se presenta a edad temprana15. Livingston et al., en un metaanálisis reporta hallazgos concordantes con publicaciones previas, un OR de 3,67 (IC de 1,18 a 11,45) para aparición de linfadenopatías cuando el LES se inicia en la juventud16.
En los casos en los cuales la linfadenopatía se presenta como la manifestación primaria del lupus casi siempre se acompañan de síntomas constitucionales adicionales, sobre todo si se trata de pacientes jóvenes17 y su presencia se ha asociado a mayor actividad de la enfermedad. Otros hallazgos encontrados en esta forma de presentación son la fatiga, fiebre, pérdida de peso, lesiones cutáneas, hepatomegalia, esplenomegalia, anticuerpos anti-DNA incrementados y niveles disminuidos de complemento5.
Típicamente los ganglios son suaves, no dolorosos, de tamaño variable entre 0,5 hasta 3 a 4cm y se presentan con mayor frecuencia a nivel cervical, axilar e inguinal4. Durante la remisión de la enfermedad las linfadenopatías tienden a desaparecer, sin embargo, en algunos pacientes disminuyen de tamaño y pueden persistentemente ser localizadas.
Los hallazgos histológicos son usualmente inespecíficos y consisten en hiperplasia folicular moderada asociada a incremento de la vasculatura o focos de necrosis con células blastoides, restos de cariorrexis, macrófagos e histiocitos18. En el lupus, las adenopatías son un hallazgo benigno y se pueden ver en cualquier fase de la enfermedad; esta apariencia en la biopsia no es específica para algún diagnóstico y puede ocurrir en una gran variedad de condiciones, incluyendo infecciones virales y otras enfermedades autoinmunes19. Los cuerpos de hematoxilina son considerados característicos de adenopatías lúpicas pero no siempre se encuentran18,20. La biopsia se podría realizar con aguja fina, sin embargo, requiere de evaluación de un patólogo experimentado; de lo contrario, sería mejor biopsia excisional y estudio con coloraciones e inmunohistoquímica, ya que el diagnóstico de linfoma no puede ser excluido inicialmente12.
Como diagnóstico diferencial se deben descartar otras condiciones con sustrato autoinmune tales como: artritis reumatoide, síndrome de Sjögren, sarcoidosis y enfermedad de Still1.
La gran mayoría de las enfermedades infecciosas se puede manifestar con linfadenopatías regionales o diseminadas, las que más frecuentemente generan este hallazgo son las infecciones bacterianas, la infección por virus de inmunodeficiencia humana, citomegalovirus, tuberculosis, infección por virus de Epstein Bar y toxoplasmosis21. Hay otras enfermedades no neoplásicas ni infecciosas que generan linfadenopatías, entre ellas la hiperplasia reactiva linfoide, la linfadenitis dermatopática, enfermedad de Rosai-Dorfman, enfermedad de Kimura, enfermedad de Kikuchi-Fujimoto y linfadenopatías asociadas a desórdenes autoinmunes y metabólicos, por lo que un abordaje adecuado del paciente que ingresa con este síndrome clínico obliga a descartar estas condiciones u otras22 que se enumeran en la tabla 32.
Causa de adenopatías. Acrónimo «MIAMI»
| Malignidad | Infecciones | Autoinmune | Miscelánea | Iatrogénica |
|---|---|---|---|---|
| LinfomasLeucemiasNeoplasias pielSarcoma KaposiMetástasis | BrucelosisEnfermedad arañazo del gatoCitomegalovirusMononucleosis infecciosaLinfogranuloma venéreoFaringitisRubeolaTularemiaFiebre tifoideaSífilisHepatitis virales | LESArtritis reumatoideSíndrome de Sjögren | Enfermedad de KawasakiSarcoidosis | Enfermedad del sueroMedicamentos |
Adaptado y modificado de Bazemore et al.2
Otra patología, no menos importante, es la linfadenitis de Kikuchi, también conocida como linfadenitis necrosante, la cual es un síndrome benigno de causa desconocida que ocurre, principalmente, en adultos jóvenes quienes presentan linfadenopatías y fiebre. La linfadenopatía típicamente es limitada a nodos cervicales y en raros casos hay linfadenopatías generalizadas. Las manifestaciones cutáneas han sido descritas hasta en el 40% de los casos. El examen patológico de los ganglios revela un patrón característico de linfadenitis necrosante. La resolución de esta linfadenitis ocurre espontáneamente y usualmente unas semanas después del inicio de los síntomas23. En ocasiones es un reto diagnóstico ya que puede simular LES con compromiso hematológico o presentarse de forma simultánea con el mismo, generando así una dificultad en la caracterización y el diagnóstico24.
ConclusiónLas adenopatías generalizadas son una condición clínica frecuente, que implica un análisis exhaustivo de todos los elementos de la historia clínica para de esta forma llegar a una aproximación razonable y lógica.
A pesar de que las adenopatías no son la manifestación más frecuente del LES y no están incluidas en los últimos criterios de clasificación, siempre deben tenerse en cuenta dentro del amplio diagnóstico diferencial, ya que un diagnóstico oportuno permite ofrecer al paciente un manejo temprano. En estas condiciones no se puede desconocer la probabilidad de concomitancia con otras enfermedades que requieran un manejo adicional; por lo que el juicio clínico y una integración de los hallazgos clínicos, patológicos y radiológicos, son esenciales para llegar a un diagnóstico adecuado.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores han obtenido el consentimiento informado de los pacientes y/o sujetos referidos en el artículo. Este documento obra en poder del autor de correspondencia.
Conflicto de interesesLos autores declaran no tener ningún conflicto de interés.
