








La enfermedad de Behçet es una entidad clínica autoinflamatoria, de etiología desconocida, generalmente con compromiso sistémico, con un patrón de exacerbación y remisión frecuente que se asocia a retraso en el diagnóstico.
El diagnóstico de esta enfermedad es complejo, por esta razón presentamos 4 casos de pacientes con enfermedad de Behçet, que durante el abordaje clínico fueron consideradas otras enfermedades de naturaleza autoinmune. La revisión integrada de la historia clínica, la aparición de úlceras orales y genitales, así como el estudio de tipificación del complejo mayor de histocompatibilidad (HLA) permitieron diagnosticar la enfermedad de Behçet.
Behçet disease is a rare autoinflammatory disorder of unknown aetiology and is characterised by systemic manifestations with an exacerbation-remission pattern, often associated with diagnostic delay.
The diagnostic approach to this disease is complex. A report is given on four cases of patients fulfilling the diagnostic criteria for Behçet disease. Other autoimmune rheumatic diseases were considered in the clinical approach. A meticulous clinical evaluation, taking into consideration relapsing aphthous ulcers in oral mucosa and genitalia, and HLA typing allowed a proper diagnosis of Behçet disease to be made.
La enfermedad de Behçet (EB) o también llamado síndrome de Behçet, es una entidad clínica autoinflamatoria de naturaleza crónica y etiología desconocida, clasificada dentro del grupo de las vasculitis de afectación de vaso sanguíneo de calibre variable no asociada a ANCA1. Se caracteriza por compromiso sistémico predominantemente mucocutáneo, articular y ocular2. La edad promedio de aparición es entre los 20-40 años de edad y no existe predilección de sexo3. La EB tiene un curso clínico diverso en la mayoría de los pacientes, con un patrón de exacerbación y remisión4, lo cual se suele asociar a retraso en el diagnóstico que, en general, supera los 5 años según lo reportado en diversas cohortes internacionales5.
La prevalencia de la EB varía en función de la localización geográfica, siendo más frecuente en los países que hacen parte de la denominada Ruta de la Seda, desde el mediterráneo hasta el extremo este de Asia6. Se ha identificado la existencia de predisposición genética asociada a HLA, que facilita la presentación y diagnóstico7. Dentro de estos, destaca el HLA-B51, del cual se han descrito más de 200 polimorfismos8–10. La frecuencia más alta ha sido reportada en Turquía con aproximadamente 420 enfermos por 100.000 habitantes, seguido de Israel, Japón, Corea y China11. En Colombia, la prevalencia de la EB es de 1,10 enfermos por 100.000 habitantes12.
La diversidad del espectro clínico, el carácter asincrónico de las manifestaciones clínicas, el no cumplimiento de la totalidad de los criterios clasificatorios así como el patrón clínico de exacerbación y remisión que caracteriza esta entidad, dificulta el diagnóstico de la EB13, convirtiéndolo en un reto clínico y generando aproximaciones terapéuticas poco eficaces14. Desde el año 2010, la EB es considerada en Colombia una enfermedad huérfana y, por consiguiente, una entidad de especial interés para el campo de la reumatología local. Con esta serie de casos, se pretende aportar en el conocimiento del comportamiento de la EB en nuestro medio dando a conocer datos clínicos de relevancia que permitan al clínico un acercamiento diagnóstico más oportuno.
Materiales y métodosSe realizó un estudio descriptivo, retrospectivo, de una serie de casos, que incluyó a 4 pacientes ambulatorios y no consecutivos con diagnóstico de EB, los cuales fueron atendidos en la consulta externa de reumatología del Hospital Militar Central de Bogotá, Colombia. Se revisaron las historias clínicas y se recabó la información en un formulario previamente establecido. Las variables sociodemográficas y relacionadas con la enfermedad, que fueron recolectadas son: edad, sexo, síntoma debutante, compromiso orgánico que indicó EB, terapia inmunosupresora prescrita, diagnósticos diferenciales, estudios de laboratorio (imágenes de radiología, perfil de autoanticuerpos, estudio genético de HLA y reactantes de fase aguda) y tiempo en años entre el inicio de los síntomas y el diagnóstico de la EB. El análisis de los resultados se hizo con estadística descriptiva, utilizando las herramientas de la hoja de cálculo de Microsoft Excel 2013.
Descripción de la serie de casosCaso 1Mujer de 37 años, enfermera de profesión, quien debutó con cuadro clínico a los 23 años, consistente en fatiga, poliartritis, dolor glúteo izquierdo irradiado a cadera ipsilateral, eritema malar, fotosensibilidad y caída del cabello. No refirió antecedentes personales y familiares ni síntomas adicionales a la revisión por sistemas. Por cuadro descrito, la sospecha diagnóstica inicial fue de LES. El perfil inmunológico documentó: ANA 1:160 patrón homogéneo, anti-DNA negativo, C3-C4 normales y factor reumatoide (FR) negativo, los reactantes de fase aguda inicialmente fueron negativos y posteriormente permanecieron levemente elevados. Se decidió tratamiento combinado con glucocorticoides sistémicos (GCS) a altas dosis e inmunosupresores (metotrexate, azatioprina e hidroxicloroquina) sin presentar mejoría. Cuatro años después, presentó úlceras orales dolorosas recurrentes, mialgias y cefalea con signos de alarma. Dentro del abordaje diagnóstico, se realizó resonancia magnética nuclear (RMN) cerebral que documentó aumento del tejido periorbitario bilateral y exoftalmos de predominio izquierdo. Durante la punción lumbar, se demostró presión de apertura 40cm de H20 y líquido cefalorraquídeo (LCR) de características normales. Se diagnosticó hipertensión intracraneana idiopática (HII) que mejoró con el drenaje del LCR. Durante los siguientes 10 años, la paciente presentó episodios de exacerbación y remisión de los síntomas mucocutáneos y articulares. Por persistencia del dolor glúteo, se realizó RMN de articulaciones sacroilíacas la cual demostró edema óseo subcondral y erosiones en la articulación sacroilíaca izquierda compatible con sacroilitis (fig. 1). A la edad de 37 años, presentó múltiples episodios de úlceras genitales (fig. 2); la sospecha diagnóstica fue enfermedad de transmisión sexual por lo que se le realizaron estudios serológicos de VIH, VDRL e IgM/IgG para herpes virus con resultados negativos. Un año después, es valorada por el servicio de reumatología. Teniendo en cuenta los antecedentes de úlceras orales y genitales, sacroilitis, artritis, HII y síntomas constitucionales, se sospechó EB. Se realizó prueba de patergia que fue positiva y tipificación de HLA documentándose: HLA-A*01,*24, HLA-B*35,*38 y HLA-DR*04;*13. Con todo lo anterior, se confirmó diagnóstico de EB tras 15 años de evolución de síntomas. Se dio tratamiento inicial con colchicina 0,5mg cada 8 h, diclofenaco 50mg cada 12 h por 3 meses sin ninguna mejoría, por lo cual se indicó adalimumab logrando respuesta clínica favorable.
 Secuencia T1. Se observan erosiones en la articulación sacroilíaca izquierda (flecha). B) Secuencia STIR. Se observa edema óseo subcondral (flecha) en los cuadrantes superior e inferior del sacro izquierdo.")

Mujer de 48 años, enfermera de profesión. Inicia síntomas a los 30 años, consistentes en fatiga, cefalea y poliartritis. Como único antecedente, la paciente es hipertensa y a la revisión por sistemas no refirió ningún síntoma adicional. Fue evaluada por reumatología y se inició abordaje por sospecha de artritis autoinmune. Se solicitó perfil inmunológico: ANA, anti-DNA, ENA, anti CCP y FR reportados negativos, los reactantes de fase aguda VSG y PCR fueron negativos, ante la negatividad de las pruebas de laboratorio descritas, se consideró que el cuadro era secundario a fibromialgia indicándose tratamiento con neuromoduladores (gabapentina, pregabalina, amitriptilina) sin mejoría de los síntomas. A los 33 años, presentó por primera vez úlceras orales recurrentes, dándose como diagnóstico en ese momento gingivoestomatitis infecciosa. En los 15 años siguientes, los síntomas articulares y mucocutáneos se presentaron de manera intermitente y a la edad de 48 años debutó con dolor lumbar inflamatorio. En vista del nuevo síntoma, reumatología realizó un nuevo abordaje diagnóstico. Dentro de los estudios: tipificación de HLA se documentó HLA-B27 negativo y se encontró HLA-B51 positivo y RMN de articulaciones sacroilíacas que documentó edema óseo subcondral en la articulación sacroilíaca derecha compatible con sacroilitis (fig. 3). Con los antecedentes de poliartritis, sacroilitis, úlceras orales recurrentes y positividad para HLA-B51 y tras 18 años de inicio de síntomas, se confirmó diagnóstico de EB. La prueba de patergia fue negativa. En el momento la paciente se encuentra en terapia con AINE con mejoría parcial, está en proceso de inicio de terapia biológica.
Caso 3 A) coronal B) axial.")
Mujer de 49 años de profesión publicista, sin antecedentes personales de importancia. A los 10 años, presentó dolor poliarticular, episodios febriles intermitentes y úlceras orales y genitales recurrentes. Fue valorada por los servicios de ginecología y dermatología sin concluirse un diagnóstico definitivo. A la edad de 20 años los síntomas remitieron espontáneamente y permaneció asintomática por 26 años. A los 46 años, la paciente presentó episodio de infección probable por virus Zika y reaparecieron los síntomas de artritis, fatiga y las úlceras orales y genitales (fig. 4). La afección articular comprometió carpos, metacarpofalángicas, codos, hombros, espalda baja y rodillas. Fue remitida al servicio de reumatología y por cuadro descrito se inició abordaje diagnóstico de enfermedad autoinmune tipo LES vs. artritis inflamatoria. Dentro de los estudios realizados destacan: anti-DNA, ANA, ENA, FR y anti CCP negativos; RNMSI fueron compatibles con sacroilitis bilateral; y tipificación de HLA que documentó HLA-A*02;*24, HLA-B*35;*51, HLA-DR*04;*16. Con base en el cuadro clínico y estudios de laboratorio descritos, después de 39 años de inicio de los síntomas se confirmó diagnóstico de EB. Se prescribieron GCS (deflazacort 6mg día) y colchicina 0,5mg tab cada 8 h durante 12 meses, sin lograr respuesta clínica favorable, por lo cual se decidió inicio de terapia biológica. Inicialmente recibió etanercept el cual fue suspendido por reacción alérgica cutánea grave; se sustituyó por adalimumab con el cual lleva 16 meses de tratamiento logrando respuesta clínica favorable.
Caso 4
Mujer de 30 años, médica de profesión quien inició síntomas a la edad de 16 años. Los antecedentes personales y familiares y la revisión por sistemas eran negativos. Cuadro clínico debutante caracterizado por: fatiga, artritis de metacarpofalángicas, interfalángicas proximales y carpos, alopecia areata, úlceras orales de características herpetiformes y pérdida involuntaria de peso. Los síntomas aparecieron de forma intermitente y asincrónica. Como único hallazgo paraclínico anormal se encontró elevación persistente de la VSG. El perfil inmunológico: ANA, ENA y FR fue reportado negativo en varias ocasiones. Recibió tratamiento con ciclos de AINE con mejoría parcial sin establecerse un diagnóstico definitivo. Fue valorada por medicina interna a la edad de 20 años y con base en el cuadro clínico y exámenes de laboratorio descritos, concluye diagnóstico de enfermedad indiferenciada del tejido conectivo. Le fue prescrito GCS e hidroxicloroquina, con leve mejoría de la sintomatología. Cuatro años después, presentó cuadro agudo de amaurosis izquierda secundaria a neuritis óptica posquiasmática, según concepto de oftalmología. Fue valorada por primera vez por reumatología y aún bajo presunción diagnóstica de enfermedad indiferenciada del tejido conectivo, se indicaron GCS a dosis altas, logrando solo recuperación parcial de la visión. Se toman nuevamente exámenes de laboratorio por sospecha de enfermedad autoinmune: ANA, ENA, anti-DNA y perfil de síndrome de anticuerpos antifosfolípido fueron reportados negativos. Se continuó tratamiento con dosis altas de GCS, sin embargo, no presentó mejoría clínica de la artritis de manos, rodillas y tobillos, y persistían úlceras orales recurrentes. Dos años después, presentó un segundo episodio de neuritis óptica izquierda de características similares al episodio anterior requiriendo nuevamente tratamiento con pulsos de metilprednisolona. Un año después, aparecieron úlceras genitales dolorosas ubicadas en labios mayores y vulva. Se descartaron enfermedades de transmisión sexual y no se modificó esquema terapéutico. Teniendo en cuenta los antecedentes de poliartritis, úlceras orales y genitales recurrentes, síntomas constitucionales, neuritis óptica y perfil inmunológico negativo, reumatología confirmó diagnóstico de EB después de 14 años de inicio de síntomas. Se adicionó al tratamiento azatioprina y colchicina durante 12 meses logrando remisión parcial de los síntomas. Posteriormente tuvo empeoramiento clínico por lo cual se prescribe adalimumab con el que lleva 17 meses presentando buena respuesta clínica.
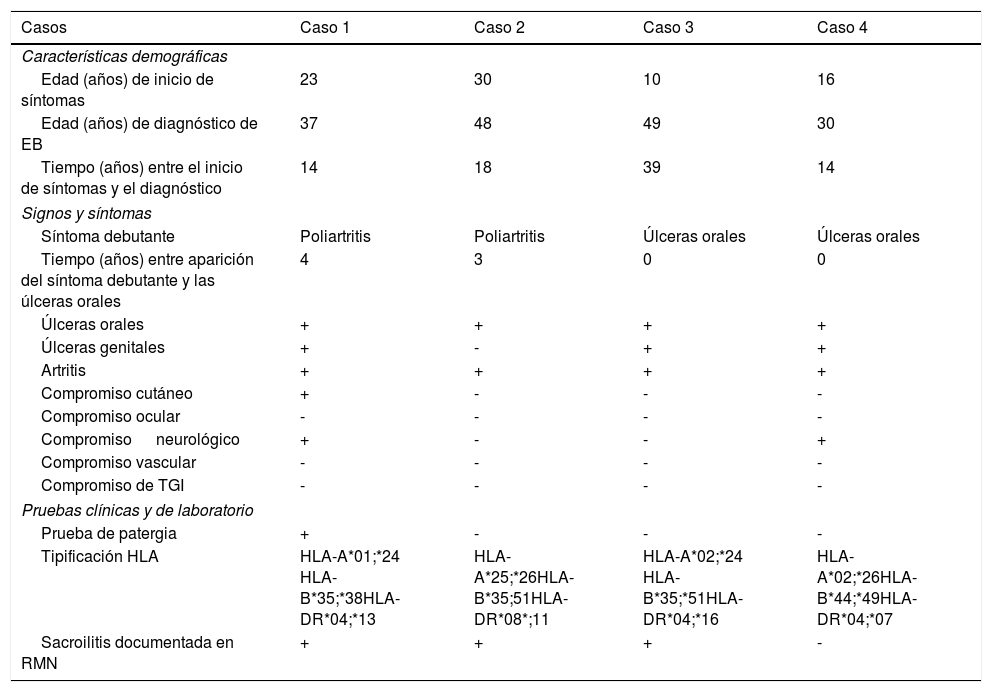
La descripción de las características demográficas y hallazgos clínicos y de laboratorio de las 4 pacientes se presenta en la tabla 1.
Descripción de características demográficas y hallazgos clínicos y de laboratorio
| Casos | Caso 1 | Caso 2 | Caso 3 | Caso 4 |
|---|---|---|---|---|
| Características demográficas | ||||
| Edad (años) de inicio de síntomas | 23 | 30 | 10 | 16 |
| Edad (años) de diagnóstico de EB | 37 | 48 | 49 | 30 |
| Tiempo (años) entre el inicio de síntomas y el diagnóstico | 14 | 18 | 39 | 14 |
| Signos y síntomas | ||||
| Síntoma debutante | Poliartritis | Poliartritis | Úlceras orales | Úlceras orales |
| Tiempo (años) entre aparición del síntoma debutante y las úlceras orales | 4 | 3 | 0 | 0 |
| Úlceras orales | + | + | + | + |
| Úlceras genitales | + | - | + | + |
| Artritis | + | + | + | + |
| Compromiso cutáneo | + | - | - | - |
| Compromiso ocular | - | - | - | - |
| Compromiso neurológico | + | - | - | + |
| Compromiso vascular | - | - | - | - |
| Compromiso de TGI | - | - | - | - |
| Pruebas clínicas y de laboratorio | ||||
| Prueba de patergia | + | - | - | - |
| Tipificación HLA | HLA-A*01;*24 HLA-B*35;*38HLA-DR*04;*13 | HLA-A*25;*26HLA-B*35;51HLA-DR*08*;11 | HLA-A*02;*24 HLA-B*35;*51HLA-DR*04;*16 | HLA-A*02;*26HLA-B*44;*49HLA-DR*04;*07 |
| Sacroilitis documentada en RMN | + | + | + | - |
Se presenta una serie de 4 casos de pacientes con EB en los cuales los síntomas más relevantes fueron: artritis, úlceras orales y genitales y constitucionales. Hubo predilección de presentación por el sexo femenino (100%) y la edad promedio de diagnóstico fue de 40,7 años. Estos hallazgos son concordantes con el estudio de aproximación de la prevalencia de EB en Colombia en el que se identificaron 523 casos documentándose relación mujer: hombre de 2,15:1 y mayor prevalencia de enfermedad entre los 45 a 49 años. En cuanto a la distribución de presentación por sexo, en la mayoría de los estudios a escala mundial se observan frecuencias similares en ambos grupos pero, en países latinoamericanos (Brasil y Colombia)12,15 se ha encontrado predilección por el sexo femenino (68% para Colombia). Es probable que este fenómeno esté relacionado con la etnicidad de la población latinoamericana y se requerirán estudios adicionales para probar esta teoría.
En 3 de las 4 pacientes, la sospecha clínica inicial fue de LES y se consideraron otras entidades como AR y espondiloartritis (EspA). La diversidad del espectro clínico, la asincronía en la presentación de las manifestaciones y la baja prevalencia de la EB dificultaron el abordaje clínico, resultando en un tiempo promedio de retraso diagnóstico de 21,5 años y en todos los casos siendo dictaminado por un reumatólogo.
En poblaciones de mayor prevalencia para EB, el tiempo de retraso diagnóstico descrito es de 5,32 ± 6,3 y de 4,54 ± 4,07 años, en un estudio multicéntrico europeo y en una población griega, respectivamente5,16. En Colombia, Toro-Giraldo et al. describieron en el 2009 una serie de 20 casos de EB y encontraron que el tiempo promedio entre la primera manifestación y el cumplimiento de los criterios diagnósticos fue de 4,45 ± 4,19 años17. En esta serie de casos, el tiempo de retraso diagnóstico es significativamente superior al descrito en otras cohortes nacionales e internacionales, poniendo de manifiesto la falta de sospecha clínica de esta entidad. Son factores que pudieron contribuir a este fenómeno: 1) tiempo prolongado de remisión de síntomas 2) valoración tardía por el servicio de reumatología y 3) tiempo prologando entre el síntoma debutante y la aparición de las úlceras orales.
Aspectos epidemiológicosEn Colombia, un estudio de aproximación de la prevalencia de enfermedades reumáticas, basado en el registro en enfermedades reumáticas del Sistema Integral de Información de la Protección Social (SISPRO) entre el 2012 y 2016, estimó una prevalencia de 0,001% (prevalencia estimada de 1,10/100.000 habitantes y de 1,49/100.000 habitantes en mayores de 18 años)12.
A escala latinoamericana, no existen estudios sobre la prevalencia de la EB; sin embargo, hay notoria diferencia entre la prevalencia local y la evaluada en países de la Ruta de la Seda donde esta es significativamente mayor: Turquía (20 – 420/100.000), Israel (120/100.000), Irak (17/100.000) y Japón (15/100.000) y en menor proporción, el norte de Europa (Alemania 2,26/100.000), Estados Unidos (0,3-6,6/100.000) e Inglaterra (0,64/100.000)4. Este comportamiento epidemiológico descrito guarda relación con la existencia de una predisposición de carácter genético que facilita la presentación y el diagnóstico de la EB7.
Curso clínicoDesde el punto de vista clínico, resalta la heterogeneidad del cuadro. Las manifestaciones más frecuentes son: úlceras orales (90-100%), úlceras genitales (85%), compromiso cutáneo (85%), ocular (50-70%), articular (30-50%), gastrointestinal (11%), de SNC (5-10%) y vascular (4%)11. En población colombiana, las descripciones de series de casos documentan hallazgos similares siendo las manifestaciones clínicas más frecuentes en todos los casos la afectación mucocutánea, ocular y articular18.
Los diagnósticos diferenciales de mayor relevancia clínica para la EB son: LES14,19, AR y EspA20. Esto se debe, en gran medida, a que estas patologías comparten síntomas articulares, mucocutáneos y oculares similares, y además son más prevalentes que la EB en nuestro medio21.
Compromiso mucocutáneo. Una característica clínica clásica en la EB son las úlceras o aftas orales y genitales presentes casi en el 100% de los pacientes para la localización oral y 85% para la genital, convirtiéndose en un sello clínico distintivo de esta entidad y, por tanto, constituyen 2 de los criterios diagnósticos mayores, respectivamente6. Las úlceras orales son el síntoma inicial en el 76% de los casos y deben recurrir por lo menos 3 veces en un año para ser consideradas manifestación de esta patología4,22. Se describen clásicamente como lesiones elevadas eritematosas que tras uno o dos días de su aparición se ulceran. Las úlceras orales se localizan en mucosa bucal, lengua, labios y encías (menos frecuente en paladar, faringe posterior y amígdalas) y las genitales en escroto, pene, región perianal, vagina o vulva23,24.
Clínicamente, las úlceras se describen como lesiones pequeñas (<10mm) de contorno redondeado u oval, superficiales y cubiertas por una pseudomenbrana blancoamarillenta18,25. Las úlceras genitales son macroscópicamente similares a las orales, pero en general, son más grandes, profundas y dolorosas, y toman más tiempo para remitir que las orales26. Otras manifestaciones cutáneas diferentes a las úlceras orales/genitales de relevancia clínica para resaltar en la EB son: lesiones papulopustulares o pseudofoliculitis (acneiformes), eritema nodoso, tromboflebitis superficial, úlceras cutáneas o nódulos, lesiones similares a celulitis y pioderma gangrenoso y lesiones cutáneas vasculíticas variadas25.
De los casos expuestos, todas las pacientes presentaron úlceras orales y 3 de 4, úlceras genitales. Ninguna de las pacientes presentó síntomas cutáneos diferentes a las úlceras. A diferencia del LES y la EspA, las úlceras en EB son dolorosas, no dejan cicatriz y remiten en un periodo de 1 a 2 semanas. Si bien en LES y EspA son un síntoma de importancia clínica, la frecuencia de presentación es significativamente menor que en EB (28%-52% para LES y 14,5% para EspA, respectivamente)27–29.
Compromiso articular. En la EB, el compromiso clásico es oligoarticular inflamatorio no erosivo, simétrico o asimétrico, intermitente y de grandes articulaciones (rodillas, tobillos, codos y muñecas)11. Con menor frecuencia, se reportan hallazgos de entesitis, sacroilitis, daño erosivo con pérdida del cartílago, formación de pannus y osteonecrosis con múltiples lesiones osteolíticas reversibles4. En los casos presentados, a 3 de las 4 pacientes le fue documentado sacroilitis, hallazgo concordante con lo descrito en otras cohortes30. En cambio, en LES y AR la clínica es poliarticular, periférica, clásicamente de pequeñas articulaciones, simétrica y además se caracterizan por la positividad de anticuerpos específicos para cada una (ANA, anti-DNA, anti-Sm, anti-nucleosoma y anti-C1q para LES y FR y anti-CCP para AR)31. La EB se diferencia de la EspA, desde el punto de vista articular, porque en la EspA la localización del dolor es típicamente axial o axial/periférico y existe asociación genética claramente definida con el HLA-B27 y HLA-B1532.
Compromiso ocular. Presente entre el 50-70% y constituye un criterio diagnóstico mayor de esta entidad. Puede comprometer cualquier estructura ocular predominando la afección de la cámara posterior del ojo por lo cual puede causar pérdida de visión irreversible33. El compromiso es bilateral en el 90% de los casos y es más frecuente y grave en hombres. Clínicamente se manifiesta con dolor ocular, visión borrosa y pérdida completa o parcial de la visión. La patología ocular relacionada con la EB característica es la uveítis y otras menos frecuentes son: vasculitis retiniana, iridociclitis, viteritis y papilitis34. En otras entidades de naturaleza autoinmune como LES y AR, el compromiso ocular es menos frecuente (30% y 15-20%, respectivamente) y la patología oftalmológica más común es queratoconjuntivitis sicca para ambas entidades35–37. Para EspA, la prevalencia de compromiso ocular es mayor que en LES y AR, siendo la espondilitis anquilosante (EA) el subtipo de EspA con mayor afectación ocular (uveítis anterior en el 25%)38. En los casos presentados, ninguna de las pacientes tuvo compromiso ocular. La neuritis óptica de la paciente del caso 4 se considerará una manifestación neurológica.
Compromiso de otros sistemas en la EB. La EB es una vasculitis multisistémica y por tanto puede potencialmente afectar cualquier sistema del organismo. Los de mayor relevancia clínica por la morbimortalidad que acarrean son: compromiso neurológico (5-10%), vascular (2-37%) y gastrointestinal (3-26%)6,13,39.
Compromiso neurológico. Puede afectarse el sistema nervioso central (SNC) y el sistema nervioso periférico (poco frecuente). El compromiso del SNC puede ser clasificado en parenquimatoso (80%) y no parenquimatoso (20%)40,41. El primero incluye: lesiones extensas de tallo cerebral/ganglios basales (la atrofia aislada del tallo cerebral es casi patognomónica de la EB), manifestaciones hemisféricas, lesiones de la médula espinal y meningoencefalitis aséptica. Clínicamente se manifiesta con signos piramidales, hemiparesia, cambios comportamentales/cognitivos, incontinencia esfinteriana y disfunción sexual40. La neuritis óptica, manifestación presentada por la paciente del caso 4, así como los síntomas sensitivos y el compromiso de la médula espinal son manifestaciones raras y poco descritas en la literatura4. Por su parte, el compromiso no parenquimatoso corresponde a oclusión o aneurismas arteriales y trombosis del seno dural resultante en HII42–45. La paciente del primer caso presentó un cuadro confirmado de HII; si bien no le fue documentado objetivamente trombosis del seno dural, las características clínicas y del LCR corresponderían a un compromiso del SNC no parenquimatoso secundario a EB.
Compromiso vascular. La EB puede afectar tanto el sistema arterial y venoso, y las lesiones vasculares prototipo más importantes son: oclusión arterial, aneurismas, oclusión venosa y trombosis. Es más frecuente en hombres jóvenes y la afección del sistema venoso (80%) es más frecuente en comparación con la arterial (20%)46. La trombosis venosa profunda de extremidades inferiores es la manifestación vascular más frecuente. Los aneurismas de la arteria pulmonar constituyen la complicación de mayor mortalidad descrita en EB4.
Compromiso del tracto gastrointestinal (TGI). Se caracteriza por inflamación de la mucosa que resulta en la formación de úlceras únicas o múltiples a lo largo de toda la extensión del mismo, siendo más frecuentes en la región ileocecal13. Clínicamente, puede manifestarse con dolor abdominal, disentería o hematoquezia o dolor abdominal agudo por perforación visceral. El diagnóstico diferencial debe hacerse con la enfermedad de Crohn pudiendo ser indistinguible en ausencia de manifestaciones extraintestinales6.
Como se ha expuesto, no existe un signo o síntoma patognomónico en la EB. La epidemiología local en términos de manifestaciones clínicas es concordante a lo encontrado en otras poblaciones. Debido a la variabilidad en el espectro clínico y la presentación asincrónica de las manifestaciones, el diagnóstico de la EB precisa de un juicio clínico detallado pues no se dispone de pruebas histopatológicas o de laboratorio diagnósticas definitivas. Los criterios de diagnóstico de la EB se anotan en la tabla 2.
Criterios internacionales para el diagnóstico de enfermedad de Behçet (ICBD) 2014
| Úlceras oralesÚlceras menores, mayores o herpetiformes observadas por el médico o el paciente, con un mínimo de 3 episodios durante un período de 12 meses | 2 puntos |
| Úlceras genitalesÚlceras o cicatrices aftosas en zonas genitales observadas por el médico o paciente | 2 puntos |
| Manifestaciones ocularesUveítis anterior y posterior o vasculitis retiniana diagnosticada por el oftalmólogo | 2 puntos |
| Manifestaciones cutáneasEritema nodoso, foliculitis, lesiones papulopustulosas o nódulos acneiformes, observados por el médico en pacientes posadolescentes no tratados con glucocorticoides | 1 punto |
| Manifestaciones vascularesTrombosis arteriales o venosas, flebitis superficiales o aneurismas | 1 punto |
| Manifestaciones neurológicasCompromiso de SNCCompromiso de sistema nervioso periférico | 1 punto |
| Prueba de patergia positiva (opcional)Hipersensibilidad cutánea caracterizada por la aparición de una pústula estéril, 24 a 48 h después de la punción cutánea con aguja, observada por un médico | 1 punto |
Sensibilidad 97%, especificidad 97% y precisión diagnóstica 97%. Puntuación ≥4 indica diagnóstico.
Fuente: International Team for the Revision of the International Criteria for Behçet's Disease (ITR-ICBD). The International Criteria for Behçet's Disease (ICBD): a collaborative study of 27 countries on the sensitivity and specificity of the new criteria. J Eur Acad Dermatol Venereol. 2014;28(3):338-47).
La prueba de patergia, incluida dentro de los criterios diagnósticos para EB, es un estudio altamente específico (98-100%) pero poco sensible (10-68%)47,48 y la positividad de la misma varía en función de la población estudiada, siendo usualmente positiva en los pacientes de países endémicos para EB, pero solo en el 20-30% de los americanos y europeos3. En nuestro medio, una prueba de patergia negativa no excluye el diagnóstico, aunque si resulta positiva, es altamente sugestiva de la enfermedad, razón por la cual se recomienda realizarse en todo paciente con sospecha clínica de esta entidad.
Ante un cuadro clínico sugestivo de EB, es preciso acudir a pruebas de apoyo diagnóstico como lo es el estudio de tipificación del HLA. Para esta entidad, la asociación más frecuentemente descrita es el alelo HLA-B51, presente hasta en el 70% de los pacientes con EB en países endémicos49,50. El HLA-B*51:01 es el subtipo con mayor fuerza de asociación a la EB. En otras poblaciones han sido implicadas otras moléculas del HLA clase I y clase II con menor frecuencia incluyendo: HLA-A*26, HLA-B*15, HLA-B*27:02, HLA-B*39:01, HLA-B*52, HLA-B*57, HLA-B*5, HLA-B*38, HLA-B*35, HLA-B*56, HLA-Cw1, HLA- HLA-Cw14, HLA-Cw15, HLA-Cw16, HLA-DRB1*04 y el HLA-DRB1*077,51,52. De los casos presentados, 2 fueron positivos para HLA-B*51 y 2 para HLA-B*35, confirmando la asociación genética descrita para esta entidad.
El tratamiento de la EB es multidisciplinario, usualmente liderado por especialistas en reumatología y debe ser individualizado en función de la edad, el tipo y gravedad del órgano o sistema afectado. No se tiene estipulado una estrategia T2T (del inglés «Treat to target») y los objetivos principales son remitir los episodios de exacerbación inflamatoria y prevenir las recurrencias53. En términos generales, la piedra angular del tratamiento del episodio agudo son los GCS solos o en combinación con otros agentes inmunosupresores4. Según recomendaciones de guías EULAR del año 2018 para el tratamiento de la EB54, los glucocorticoides tópicos son la primera de línea de tratamiento para las úlceras orales y genitales. Otra alternativa es talidomida, sin embargo, con un alto porcentaje de efectos secundarios. La colchicina está indicada en artritis y prevención de manifestaciones mucocutáneas (especialmente eritema nodoso y úlceras genitales)55. En afección grave de órganos internos, están indicados los GCS a altas dosis con o sin ciclofosfamida, seguido de GCS orales de mantenimiento e inmunosupresores (azatioprina, ciclosporina). En términos generales, azatioprina tiene indicación en uveítis, trombosis venosa profunda, compromiso del TGI, del SNC y manifestaciones mucocutáneas resistentes a otros tratamientos. El uso de terapias biológicas se ha reservado en pacientes que no responden a la acción de medicamentos antirreumáticos modificadores de la enfermedad y la experiencia clínica ha sido principalmente con anti-TNFα54. La efectividad de adalimumab ha sido reportada en series de casos para el tratamiento especialmente del compromiso ocular (uveal) y menos frecuente para el mucocutáneo (úlceras genitales), TGI, SNC (vasculitis cerebral) y aneurisma bilateral de la arteria pulmonar53. En 3 de los 4 casos, se indicó tratamiento con adalimumab por falla terapéutica a GCS, inmunosupresores o colchicina, especialmente por persistencia de síntomas articulares y mucocutáneos resistentes a tratamiento convencional con una adecuada respuesta en los casos presentados.
ConclusiónLa EB es una vasculitis multisistémica, de origen desconocido y curso clínico con exacerbaciones y remisiones de duración y frecuencia impredecibles. Si bien la prevalencia estimada en nuestro medio es baja, la EB es una entidad clínica con morbilidad importante, convirtiéndola en una enfermedad de especial interés para el campo de la reumatología. La presencia de úlceras orales y genitales es una característica clínica distintiva y debe hacer sospechar la EB en el contexto de un paciente con sospecha de autoinmunidad, especialmente cuando hay concurrencia de síntomas articulares, oculares, cutáneos y neurológicos. La atrofia aislada del tallo cerebral y los aneurismas de la arteria pulmonar son manifestaciones raras pero altamente sugestivas de esta entidad. Se recomienda realizar estudio genético de tipificación de HLA y la prueba de patergia dentro del abordaje clínico. Se requieren estudios de caracterización sociodemográfica y clínica con un mayor número de pacientes en Colombia para conocer de forma más detallada el comportamiento de esta entidad en nuestro medio y acortar el tiempo de diagnóstico. En todos los casos, debido a la heterogeneidad del cuadro clínico, el abordaje de esta enfermedad debe ser multidisciplinario.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
