




Antisynthetase syndrome (AS) is an autoimmune idiopathic inflammatory myopathy (IIM) identified by the positivity of anti t-RNA synthetases. Clinical manifestations include inflammatory myopathy, interstitial lung disease (ILD), mechanic's hands (hyperkeratosis lesions of the fingers), arthritis or Raynaud's phenomenon. The objective of our study was to describe the clinical, immunological, and therapeutic characteristics of AS and to highlight its particularities compared to other IIM.
MethodsRetrospective data from the internal medicine department of Mongi Slim hospital in Tunis were collected over a period of 16years. We compared demographic, clinical, biological and morphological findings, treatments, and outcomes of patients with AS with a group of patients with IIM other than AS.
ResultsThirty patients with AS and twenty patients with other IIMs were included. Patients with AS had significantly more amyopathic forms (p=0.034) and ILD (p<0.001). Cyclophosphamide was more frequently used in the treatment of severe ILD than in that of other IIMs (p=0.023).
ConclusionAS is a rare entity with a heterogenous clinical spectrum. It is associated with increased morbidity and mortality especially if the lung involvement is severe.
El síndrome de antisintetasa (AS) es una miopatía inflamatoria autoinmune idiopática (IIM) identificada por la positividad de los anticuerpos anti-t-ARN sintetasas. Las manifestaciones clínicas incluyen miopatía inflamatoria, enfermedad pulmonar intersticial (ILD), manos de mecánico (lesiones hiperqueratósicas en los dedos), artritis o fenómeno de Raynaud. El objetivo de nuestro estudio fue describir las características clínicas, inmunológicas y terapéuticas de la AS y resaltar sus particularidades en comparación con otros IIM.
MétodosSe recopilaron datos retrospectivos del Departamento de Medicina Interna del Hospital Mongi Slim en Túnez durante un periodo de 16 años. Comparamos los hallazgos demográficos, clínicos, biológicos y morfológicos, los tratamientos y los resultados de los pacientes con AS con un grupo de pacientes con IIM diferentes al AS.
ResultadosSe incluyeron treinta pacientes con AS y veinte pacientes con otras IIM. Los pacientes con AS presentaron formas amioapáticas significativamente más frecuentes (p=0,034) y ILD (p<0,001). El ciclofosfamida se utilizó con mayor frecuencia en el tratamiento de ILD severas en comparación con otras IIM (p=0,023).
ConclusiónEl AS es una entidad rara con un espectro clínico heterogéneo. Está asociado con una mayor morbilidad y mortalidad, especialmente si la afectación pulmonar es severa.
Idiopathic inflammatory myopathies (IIM) include dermatomyositis (DM), inclusion-body myositis, immune-mediated necrotizing myopathies (IMNM) and overlap myositis (OM) with myositis including anti-synthetase syndrome (AS) [1].
All those myopathies are characterized by muscular inflammation resulting in muscle weakness, increased muscle enzymes and extra muscular manifestations [2].
AS is characterized by myositis, interstitial lung disease(ILD), cutaneous involvement, arthritis or Raynaud's phenomenon and positivity of anti t-RNA synthetases, with the anti-histidyl (anti-Jo1) being the most common of these antibodies.
The prevalence of anti-Jo1 antibodies in the general population is still unclear [3]. It is nearly 1.5 per 100,000 population [4]. The disease mainly affects adults (average age 50 years with a range of 22–74 years). Females are more prone to the syndrome than males by a ratio of 2–3:1 [1].
In patients with AS, the presence of ILD is associated with an increased mortality and morbidity, and the positivity of AS antibodies is a strong predictor for the development of ILD [5,6]. The objective of our study was to describe the clinical, immunological, and therapeutic characteristics of AS and to highlight its particularities compared to other IIM.
MethodsDemographic data, clinical, biological and morphological findings, treatments, and outcomes were retrospectively collected over a period of 16 years (from 2005 to 2021). The study was conducted in the department of Internal Medicine in Mongi Slim Hospital in Tunis, Tunisia. We included patients aged over 18 years who satisfied the criteria for AS according to Connors and al. classification [7]. We then compared the AS group (30 patients) with a second group of patients (n=20) with IIMs other than AS: DM (n=9), IMNM (n=7) and other OM (n=4). The latter responded to the IIM criteria elaborated by Troyanov et al [8].
Patients with missing or unusable data were excluded from the study.
The study was approved by the local ethical committees, and the patients were reported anonymously.
Statistical analysisThe data were imported and processed using SPSS. For quantitative variables we calculated median and interquartile range (IQR) and absolute and relative frequency with the percentage values for the qualitative ones. Since our sample did not exceed thirty patients, we applied non-parametric tests to compare medians such as the Mann–Whitney test for quantitative variables and Chi-square or Fisher exact test for categorical variables.
Significance was defined at p<0.05.
The study did not involve the use of artificial intelligence in its various stages.
ResultsA total of fifty patients with IIM were included in our study. They were divided into two groups: 30 patients with AS and 20 patients with other inflammatory myopathies. There were 25 (83.3%) female patients, and the mean age of patients was 48.1 years. The majority of patients (86.7%) did not have any prior relevant medical history and one patient had been on statin before disease onset. The first manifestation of the disease was pulmonary in 66.7% of cases, articular (13.3%), muscular (10%), cutaneous (3.3%), cardiac (3.3%) and a prolonged fever in one case. The median diagnostic delay between symptoms onset and diagnosis was 211 days.
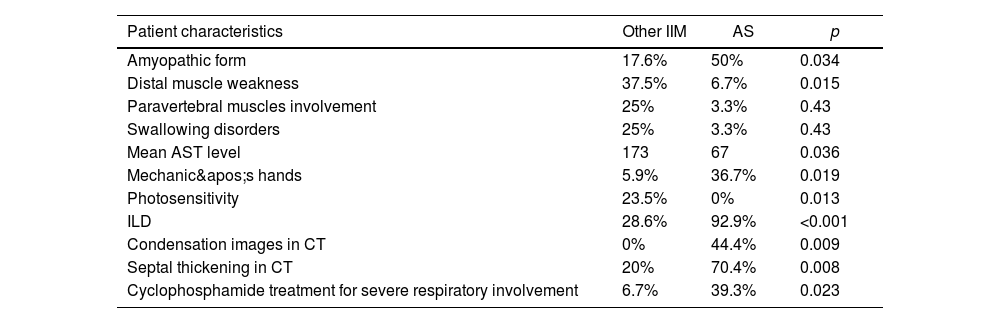
Half of our patients had an amyopathic form of AS. The muscular weakness was proximal and symmetrical in respectively 30 and 33.3% of cases. The majority of our AS patients did not have any swallowing disorders (96.7%) or any respiratory muscles involvement manifestations, while 25% of the IIM patients had swallowing disorders and 12.5% of the patients were treated for respiratory problems (p=0.43) as shown in Table 1.
Comparison between AS and other IIM patient groups.
| Patient characteristics | Other IIM | AS | p |
|---|---|---|---|
| Amyopathic form | 17.6% | 50% | 0.034 |
| Distal muscle weakness | 37.5% | 6.7% | 0.015 |
| Paravertebral muscles involvement | 25% | 3.3% | 0.43 |
| Swallowing disorders | 25% | 3.3% | 0.43 |
| Mean AST level | 173 | 67 | 0.036 |
| Mechanic's hands | 5.9% | 36.7% | 0.019 |
| Photosensitivity | 23.5% | 0% | 0.013 |
| ILD | 28.6% | 92.9% | <0.001 |
| Condensation images in CT | 0% | 44.4% | 0.009 |
| Septal thickening in CT | 20% | 70.4% | 0.008 |
| Cyclophosphamide treatment for severe respiratory involvement | 6.7% | 39.3% | 0.023 |
Mean peak creatine kinase (CK) was 1679 in AS patients and 2855 in other IIM. MRI findings were not found in our AS population, whereas 50% of the IIM patients had muscle atrophy, fatty degeneration or both.
In almost a third (28.6%) of our patients, EMG confirmed the myopathic activity. In the other cases it was normal in 23.6% of cases, showed a neurogenic trace in 14.3% of cases or showed a mixed neurogenic and myogenic activity in 23.8% of cases.
Muscular biopsy, when performed, showed a perifascicular necrosis, similar to what is seen in DM in 16.7% of cases. Perimysial monocellular infiltrates and diffuse myofiber necrosis were the most common findings (66.6%).
Cutaneous signs were present in 66.7% of cases and mechanic's hands in 36.7% of cases. Other skin lesions were rarely seen (heliotrope rash in 6.7% of cases, Gottron's sign in 3.3% of patients and skin ulcers in 3.3% of patients. Raynaud's phenomenon was observed in 33.3% of our AS patients with capillaroscopic findings suggestive of microangiopathy in all those cases.
Respiratory involvement (which was a diagnostic element in 66.7% of cases) preceded the muscular deficit in 68.4% of cases as shown in Fig. 1.

ILD occurred in 92.9% of the patients with AS, and was revealed by dyspnea (40%), coughing (6.7%), respiratory failure (23.3%) and was asymptomatic in 10% of cases.
Pulmonary hypertension was diagnosed in 30% of patients and was often asymptomatic (80%).
Main elementary radiological findings were ground glass opacities (66.7%), honeycombing (14.8%), reticulations (29.6%), nodules or micronodules, consolidation (44.4%) and septal thickening (70.4%). These lesions were predominantly peripheral and were found in the lung bases (93.3%), and were consistent with a non specific interstitial pneumonia (NSIP) aspect (33.3%) or usual interstitial pneumonia (UIP) (25.9%). Fibrosis was found in 14.8% of the CT scans.
Pulmonary function tests showed a restrictive physiology in 87% of the cases with a low mean total lung capacity (TLC) at 57.6% (<80% the predicted value).
Five patients (17.2%) were transferred to the critical care unit after their respiratory condition worsened.
Articular manifestations consisted of polyarthralgia in 62.4% of patients. Large joints were more often a source of pain more often than the smaller ones (31.7 vs 20%).
Cardiac manifestations were noted but they are rare in our series: myocarditis (3.3%), pericarditis (3.3%), coronaropathy (3.3%) and heart failure (6.7%).
One patient (3.3%) in the AS group had an associated ovarian cancer and one patient in the IIM group had a breast cancer (6.7%).
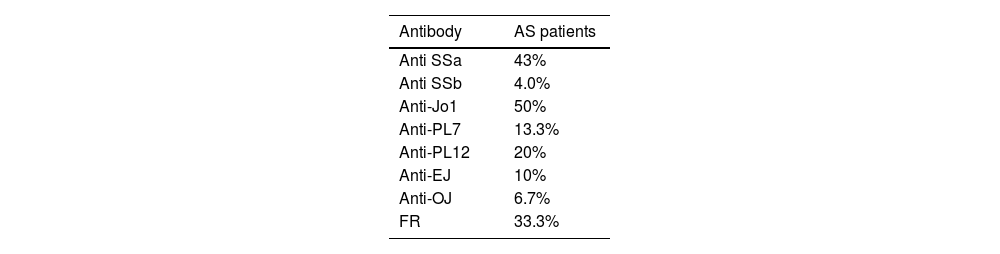
The immunological characteristics of our patients are shown in Table 2.
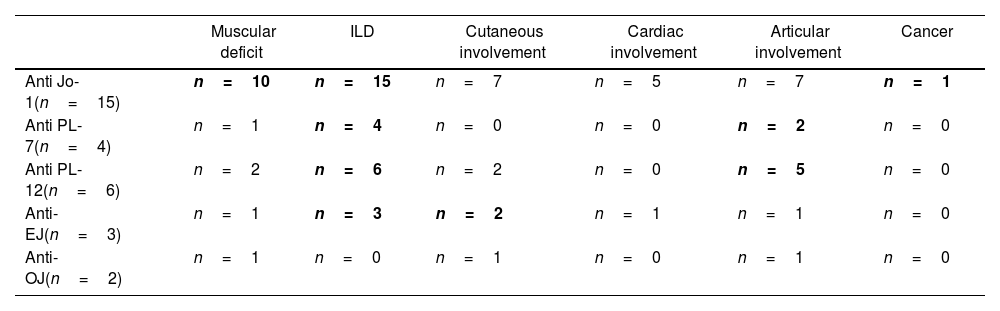
In Table 3, we have summarized the variability in clinical phenotypes according to the Myositis-specific antibody (MSA):
Clinical phenotypes according to the Myositis-specific antibody.
| Muscular deficit | ILD | Cutaneous involvement | Cardiac involvement | Articular involvement | Cancer | |
|---|---|---|---|---|---|---|
| Anti Jo-1(n=15) | n=10 | n=15 | n=7 | n=5 | n=7 | n=1 |
| Anti PL-7(n=4) | n=1 | n=4 | n=0 | n=0 | n=2 | n=0 |
| Anti PL-12(n=6) | n=2 | n=6 | n=2 | n=0 | n=5 | n=0 |
| Anti-EJ(n=3) | n=1 | n=3 | n=2 | n=1 | n=1 | n=0 |
| Anti-OJ(n=2) | n=1 | n=0 | n=1 | n=0 | n=1 | n=0 |
Seventy-two percent of our patients were started on a pulse therapy of methylprednisolone and 93.1% received 1mg/kg/day of glucocorticoids (GC). Azathioprine was the most prescribed immunosuppressant drug (68%). Methotrexate was prescribed in 32.1% of cases either as a steroid sparing agent, or in case of resistance to GC or lack of improvement with azathioprine. Over a third (39.3%) of our patients received 6 monthly perfusions of cyclophosphamide after developing severe respiratory involvement.
Cyclophosphamide was more frequently used in the treatment of severe ILD than in that of other IIMs (p=0.023). Rituximab was given as a second-line treatment in 7.1% of patients.
We obtained remission in 33.3% of the patients with muscular involvement and 50% had an early recurrence after initial regression of the deficit. ILD was resistant to treatment in 40% of AS patients.
DiscussionIn recent years, new classifications and diagnostic criteria of IIM and of its subtypes have been proposed [9]. AS can be classified in the presence of anti-synthetase antibody and one or all of the triad symptoms: myositis, arthritis and ILD [10]. However, only a minority of patients (19% in one study) [11] presented this full triad at disease onset, or even after extended follow-up [12]. Since the clinical spectrum of AS is heterogenous, it may be difficult to make the diagnosis at disease onset. An average diagnostic delay of six months was described in patients with anti-Jo-1 antibodies [12], which was almost equivalent to the mean diagnostic delay in our study (211.7 days).
EULAR/ACR recently established diagnostic criteria for IIMs and identified items permitting to differentiate IIM patients from patients with other autoimmune diseases not affecting the muscles [13]. One of the limitations of EULAR/ACR criteria is that only anti-Jo1 was included as a biological item among MSA.
AS is associated with the presence of autoantibodies to aminoacyl-transfer RNA (tRNA) synthetases which are among the MSA (e.g., anti-Jo1, anti-OJ, anti-PL7, anti-PL12, anti-EJ, anti-Ks, etc.), and which play a key role in the pathogenesis of the muscular and lung injury and directly correlate with the disease activity.
In our study, anti-Jo1 was the most commonly present antibody in AS patients, followed by anti-PL7 and anti-EJ.
Clinical characteristicsLike other IIM subtypes, AS affects females more frequently than males (female to male ratio is estimated at approximately 7:3) [14], this was supported by our own findings; we had a predominantly feminine patient population (83.3%).
MyositisAlthough classified as IIM, myositis may not necessarily be the prominent manifestation. Typically, AS patients experience weakness in the proximal muscles [15], this was the case for a third of our patients. Amyopathic onset was found in half our patients, whereas in the literature it is less typical in patients with AS [10].
A majority of our AS patients did not have any swallowing disorders (96.7%) while Nogushi et al [15]. observed them in a third of the patients.
According to Andersson et al., MRI examinations of the thigh muscle revealed abnormalities in 65% of patients with AS, but none of our patients exhibited radiological abnormalities. An important detail that may explain this difference is that we did not perform an MRI for all fifteen AS patients with a muscular deficit.
In almost a third (28.6%) of our patients, EMG confirmed the myopathic activity. EMG findings in AS are indistinguishable from those of other IIM. Characteristic findings include increased spontaneous activity as manifested by sharp waves and fibrillation, low-amplitude, short duration motor unit potentials, and early recruitment [16].
EMG and muscle MRI might be useful to monitor the disease activity and to distinguish between persistent disease activity and permanent damage to the inflamed muscles.
Some authors suggest that muscle biopsy may not be necessary in most patients with MSA [16]. The most common histological finding of muscle biopsy in AS patients is perifascicular necrosis [15]. This finding was seen in 16.7% of the performed muscle biopsies in our studies.
Muscular involvement was found in 66.7% of anti-Jo1 positive patients. It was less frequent in other subgroups. Hervier et al. also shared this finding [17].
Interstitial lung disease (ILD)In 15–40% of AS patients, ILD is the first disease manifestation [18]. In our study, the disease was revealed primarily by ILD (66.7%).
Various studies have reported the prevalence of ILD in AS to range from 69% [19] to 100% [11]. In our study, the prevalence of ILD was 92.9%.
High-resolution computed tomography (HRCT) is critical in the diagnosis and follow-up of patients with AS. The most common radiological patterns noted in patients with AS on HCRT are non-specific interstitial pneumonia (NSIP), organizing pneumonia (OP), or mixed NSIP-OP [20]. The abnormalities are usually bilateral and are either limited to the lung bases, or diffuse, or distributed in sheets. Unevenness of the pleural membrane, thickening of the septa, ground-glass densities, areas of consolidation, and linear shadows usually indicate lymphocytic alveolitis, which is classically responsive to glucocorticoid therapy in patients with connective tissue disease. Pulmonary fibrosis manifests itself as peribronchovascular thickening, Kerley B lines, reticulonodular densities, bronchiectasis, and bronchiolectasis. Honeycomb images indicate fixed irreversible fibrosis, which is usually unresponsive to glucocorticoid therapy [21].
CT scans and lung biopsies usually display NSIP in 39 to 72.5% of the analyzed patients with AS [22]. It was 33.3% in our study.
Pulmonary hypertension (present in a third of our patients) significantly worsened the prognosis, as only 58% of the patients with Asassociated pulmonary hypertension survived three years [17].
In the course of AS, ILD is more prevalent, more frequently symptomatic and persistent in anti-PL-7 and anti-PL-12-positive patients and rather less symptomatic in anti-Jo1 patients [23]. This was supported by our findings.
ArthritisPolyarthralgia and polyarthritis are the most common articular manifestations of AS, seen in 58–70% of AS patients [11,12]. Articular manifestations were found in 62.4% of the cases in our study. Joint inflammation during AS usually manifests itself as symmetrical polyarthritis and affects predominantly proximal interpharyngeal, metacarpophalangeal or wrist joints. Larger joints, such as the knees, elbows, shoulders, ankles or hips, as well as distal interpharyngeal or feet joints are less frequently involved [24].
Cutaneous signsMechanic's hands (MH), defined as fissured, scaly, non-itching hyperkeratosis, located at the palmar and lateral sides of the hand sand fingers, are one of the pathognomic signs found in AS. Its prevalence ranges from 16 to 21% [25]. It was 36.7% in our study.
More than half of AS patients develop Raynaud's (this prevalence was at 33.3% in our study), which in some cases can be severe, leading to digital ulceration [25].
Risk of neoplasmAccording to Hervier et al. the frequency of cancer in AS patients does not differ from the incidence observed in the general population [17]. Only one patient had an ovarian cancer in our cohort of patients. Screening AS patients for potential neoplasms according to individual risk is advisable nonetheless.
TreatmentGlucocorticoids (at 1mg/kg) are the first-line agent for patients with AS associated ILD. The tapering regimen is prolonged, with total duration determined by the disease course. There is no consensus on the most effective steroid-sparing immunosuppressive agent or regimen. Agents that have been used with variable success have included cyclophosphamide [26], azathioprine [27], mycophenolate mofetil [28], cyclosporine [29], tacrolimus [30], intravenous immunoglobulin [31], and rituximab [32].
Cyclophosphamide was more frequently used in our study to treat severe ILD than to treat other IIMs (39.3 vs 6.7%, p=0.023). This is in accordance with the available data [33].
LimitationsOur study was a retrospective monocentric study with a small sample size not allowing us to use multivariate analysis. EMG and muscle MRI were not easily accessible in our center.
ConclusionsAntisynthetase syndrome should be considered in patients with a wide range of manifestations, including atypical polyarthritis, pulmonary fibrosis, and mechanic's hands.
The currently available classification criteria do not fully correspond with the clinical patterns of the disease. There are important differences among the antisynthetase antibodies in their clinical manifestations, prognosis, and response to the different therapeutic options. In our study anti-Jo1 was the most commonly present antibody in AS patients, followed by anti-PL7 and anti-EJ. These findings could differ based on the population and ethnic groups investigated. ILD was more prevalent, more frequently symptomatic and persistent in anti-PL-7 and anti-PL-12-positive patients. Treatment is guided by the most severe and life- or organ-threatening disease manifestation, typically ILD.
Authors’ contribution1st author: Writing-Review and editing.
2nd author: Writing-Review and editing.
3rd author: Writing original draft.
4th author: Supervision.
5th author: Supervision.
6th author: Validation.
7th author: Validation.
8th author: Validation.
9th author: Validation.
Ethical considerationsApproved by Mongi SLIM hospital ethical committee.
FinancingNone.
Conflict of interestsNone.
