





Peanuts (Arachis hypogaea L.) are among the most important leguminous crops in Argentina. During the growing season, they are frequently attacked by fungal diseases, including Thecaphora frezii. The spores of T. frezii are structures that confer resistance to this phytopathogen. The transition from teliospore to hypha is a characteristic process of some fungi, which is essential for completing their life cycle. Using the transcriptomes of teliospores and hyphae of T. frezii, we aimed to identify genes that were differentially expressed during this transition, and we found 134 up-regulated and 66 down-regulated genes, which would participate in different cellular processes such as: (a) cell cycle and DNA processing; (b) cell fate; (c) rescue, defense and cellular virulence; (d) detoxification by CYP450; (e) energy; (f) nutrient interaction and nutritional adaptation; (g) metabolism; (g) proteins with binding functions or cofactor requirements; (h) stress, cell differentiation and biogenesis of cell components; and (i) transport, cell communication and transcription. The identification of genes in T. frezii and their expression levels during different stages of differentiation could contribute to our understanding of the biological mechanisms in this fungus.
El maní (Arachis hypogaea L.) es uno de los cultivos leguminosos más importantes de Argentina. Durante el ciclo de siembra, es atacado frecuentemente por enfermedades de origen fúngico, entre ellos por Thecaphora frezii, cuyas esporas son estructuras que le confieren resistencia a este fitopatógeno. El pasaje de teliospora a hifa es un proceso característico de algunos hongos, siendo fundamental para que el ciclo de vida se complete. A partir de los transcriptomas de las teliosporas e hifas de Thecaphora frezii nos propusimos identificar aquellos genes que estuvieran diferencialmente expresados en este pasaje y encontramos 134 up-regulados y 66 down-regulados, los cuales participarían en diferentes procesos celulares como son a) ciclo mitótico y procesamiento del ADN; b) destino de la célula; c) rescate, defensa y virulencia celular; d) desintoxicación mediada por CYP450; e) energía; f) interacción de nutrientes y adaptación nutricional; g) metabolismo; g) proteínas con funciones de unión o requerimiento de cofactores; h) estrés, diferenciación celular y biogénesis de componentes celulares; i) transporte, comunicación de las células y transcripción. La identificación de genes de Thecaphora frezii y sus niveles de expresión en distintos estadios de diferenciación podrían contribuir al conocimiento de mecanismos biológicos presentes en este hongo.
Thecaphora frezii (T. frezii) is a fungus that infects peanut plants (Arachis hypogaea L.)4 which generates brown carbonaceous masses in fruits and leads to significant economic losses in these plantations. These teliospores are resting structures that enable the fungus to overwinter and survive in the soil for years35. When peanut pegs penetrate the soil, their exudates disrupt teliospore dormancy, promoting spore germination and, thus, local infections15. This infection develops during peanut pegging. The germination of teliospores comprises the development of a probasidium, which leads to basidiospores via meiosis. Once basidiospores germinate, compatible haploid germ tubes fuse to form a mycelium, responsible for the infection. When peanut gynophores dig into the ground, dikaryotic hyphae can pass through them, colonize tissues, and substitute cells with reddish-brown teliospores. During shelling, teliospores are released and remain on the soil, where they can survive in a metabolically dormant state.
Several pathogenic fungi for plants develop this transition from spores to hyphae to exert their lifecycle, such as Ustilago maydis (U. maydis)28, Microbotryum violaceum28, Ophiostoma ulmi7, Taphrina deformans7 and Holleya sinecauda7. Note that T. frezii belongs to the same smut class as U. maydis, and recent research on this fungus shows differential gene expression during teliospore germination33 and increased protein level after 6h from teliospore germination, also along with an increase in RNA synthesis. This morphological shift, typically triggered by multiple environmental signals, is tightly controlled by complex genetic pathways to ensure successful pathogenic development. Mutation or drug inhibition of this passage has been shown to block the process of pathogenicity, and it is a clear example of the biological event of cell differentiation, representing a basic model for the study of this important phenomenon26.
In this study, we aim to identify some genes that are differentially expressed during the transition from teliospore to hyphal stage in the fungus T. frezii according to specific in vitro culture conditions. The findings obtained in this work will possibly contribute to understanding some of the biological mechanisms underlying T. frezii development, especially in this essential shift point, which gives rise to the formation of basidiospores and later hyphae, just before infective structures are formed.
Materials and methodsCollection, isolation and cultivation of Thecaphora freziiT. frezii teliospores were found in hypertrophic peanut pods, indicating the presence of the disease. Pods from plants grown in General Cabrera, Córdoba, Argentina, were superficially disinfected with 0.5% NaOCl (v/v); after that, teliospores were obtained and disinfected with 5% NaClO (v/v), washed twice with sterile distilled water, plated on potato-dextrose agar (Britannia PDA) and incubated at 26°C in the dark until germination31. Daily observation and a count of colonies in PDA plates was made over 15 days, to calculate the germination rate.
Nuclear staining of Thecaphora frezii hyphaeHyphae were stained according to Arias et al.2 Briefly, they were incubated for 24h in 70% (v/v) ethanol, then washed with sterile distilled water and then treated with RNase A (65°C, 10min). Propidium iodide (1% v/v) was added for 10min followed by two rinses with sterile distilled water. Cell nuclei were visualized under a fluorescence microscopy Zeiss – Axio observer D1 (Zeiss, Germany). The microscope was fitted with a Mono-camera AxioCam 506, and images were processed using the Imaging Software ZEN 2 Core.
RNA extractionTotal RNA was extracted from both teliospores and hyphae of T. frezii using the TRIzol reagent (Invitrogen, California, USA), following the manufacturer's instructions30 (Invitrogen, California, USA). To initiate this process, a pool of teliospores was created, harvested from carbonaceous peanut pods from 20 distinct plants. This sample was subsequently divided into two subsets. One subset was designated for RNA extraction and subsequent RNAseq analysis and the second subset was employed to cultivate mycelia on PDA plates, and RNA was later extracted from three separate plates. To ensure the purity of the RNA samples, genomic DNA was effectively eliminated through on-column digestion using DNase (Qiagen, Germany) at the recommended concentration by the manufacturer. The success of the RNA extraction process was evaluated in terms of both quality and quantity. Potential RNA degradation and impurities were assessed using a 1.5% (w/v) agarose gel, while RNA purity was confirmed using a NanoPhotometer spectrophotometer (Implen, California, USA). RNA concentration was calculated using the Qubit RNA assay kit and Qubit Fluorometer 2.0 (Life Technologies, California, USA).
Library preparation and RNA-SeqThe cDNA library was prepared using the RNA of T. frezii's teliospores and hyphae. They were performed using the NEB Next Ultra RNA kit for Illumina (Nebraska, USA) according to the manufacturer's instructions. Briefly, mRNA was purified from total RNA using poly-T oligo-attached magnetic beads. Fragmentation was carried out with divalent cations under elevated temperature in NEB Next First Strand Synthesis Reaction Buffer (5×). The first strand cDNA was synthesized using random hexamer primers and M-MuLV reverse transcriptase. The second strand cDNA synthesis was subsequently performed using DNA polymerase I and RNase H. cDNA was then size-selected and adaptor-ligated, and the products were selectively enriched. The clustering of the index-coded samples was performed on a cBot Cluster Generation System using TruSeq PE Cluster Kit v3-cBot-HS (Illumina, NEB, USA), following the manufacturer's instructions.
RNA-Seq data analysisThe cDNA libraries were sequenced on Illumina HiSeq 1500 to generate 2× 150bp pair-end reads at the INDEAR service facilities (Rosario, Argentina). Briefly, a de novo transcriptome was assembled with all reads, then the gene expression in each condition was measured with the counts per million (CPM) of reads. RNA-Seq read quality was confirmed by FastQC software (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) and compared (teliospores and hyphae). Fold change (FC) and statistical significance for all comparisons were determined via General Linear Model statistics using the EdgeR package version 3.4.2 of Bioconductor25 in the R environment (version 3.0.2, R Core Team, 2018)23.
T. frezii differentially expressed genes from teliospore to hyphal stageData obtained from RNA-Seq were used to identify differentially expressed genes, involved in the T. frezii life cycle, during the transition from teliospore to hyphae. A minimum 2-fold change up or down was the cut-off to consider whether a gene was positively or negatively differentially expressed. However, in specific cases, less than 2-fold differences were considered, particularly focusing on the different expression of certain genes that have been previously observed in other fungi.
FungiFun222, a tool for performing functional enrichment analysis of fungal genes, was used to obtain functional annotation of the differentially expressed genes of T. frezii teliospores and hyphae. As T. frezii is not present in the FungiFun2 database, we used orthologous gene sequences from U. maydis to assign functions to T. frezii genes.
qPCR analysis of selected genesFor the study of quantitative expression by real-time PCR, some of the differentially expressed genes in the transition from teliospores to hyphae were selected. The oligonucleotide list for the genes (some genes differentially expressed in other fungal species were selected) were designed using the Primer-Blast program (NCBI, NIH) (Table S1). Quantification of gene expression was performed through the real-time PCR detection system StepOne Plus Real-Time PCR system® (ThermoFisher, Massachusetts, USA).
cDNA was prepared from the same RNA samples as those used for RNA-Seq analysis, using the enzyme SuperScript® III Reverse Transcriptase (Invitrogen; Thermo Fisher Scientific™, USA) and following the manufacturer's recommendations. The qPCR was performed with three biological replicates using the Sybr® Green Master Mix kit (Applied Biosystems; Thermo Fisher Scientific, California, USA), following the manufacturer's recommendations.
Relative gene expression was performed using the actin transcript as a reference gene for expression normalization (this gene is highly used for fungal gene expression normalization20,29). The program used for all the targets was: 95°C for 3min, 40 cycles of 95°C for 10s and 60°C for 30s. Fluorescence was read after this step. At the end of the program, temperature was reduced from 95°C to 65°C at a rate ramp of 0.1°C/s, which allowed evaluation of the melting curves for each reaction. The specificity of the amplicons was verified by analyzing the melting curves and by sequencing the fragments obtained. The expression change in the target gene, relative to actin expression, was calculated using the 2−ΔΔCT method13. The mean and SE (±) were then determined for each of the different samples.
Statistical analysisStatistical analysis was conducted using the InfoStat Software24. All data were calculated as the mean±standard deviation. Heat map graphical representation was performed using Pheatmap package version 1.0.12 in RStudio (2022.07.1 Build 554, R environment version 4.2.1). Genes were grouped based on gene expression levels in a hierarchical clustering process.
Gene accession numbersAccession numbers of all genes (mRNA) used in this study are listed in Tables 1–3 and S1–S3.
ResultsIn this manuscript, a transcriptomic analysis was performed focusing on the transition from teliospores to hyphae. Total RNA from both structures of T. frezii was successfully extracted using the TRIzol protocol providing an acceptable RNA recovery yield and quality and was used to prepare the cDNA library for a comparative analysis of expression between both stages.
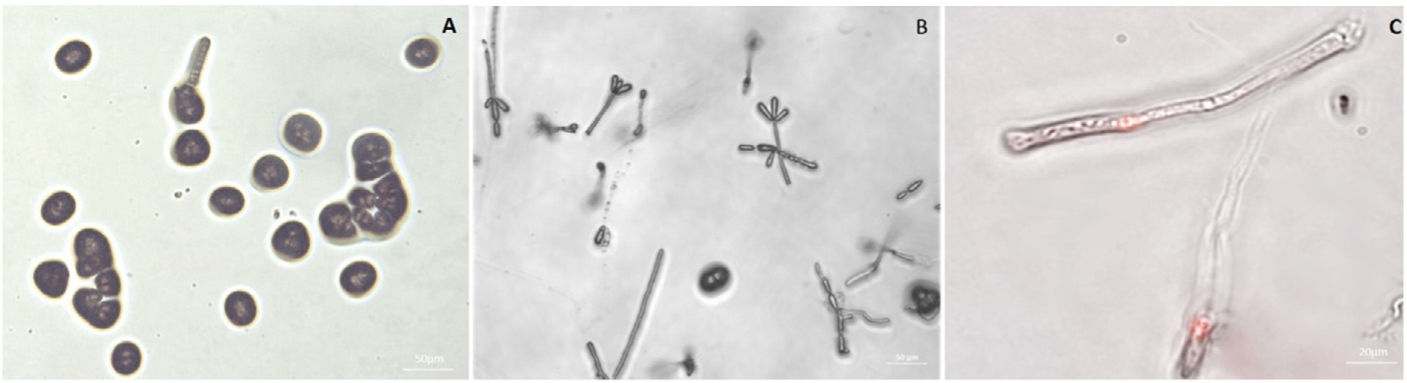
Identification of T. frezii genes involved in the transition from teliospore to hyphaeTranscription levels were compared between two T. frezii stages (teliospores and hyphae). RNA was obtained from teliospores from peanut pods with disease symptoms, and hyphae were obtained from the germination of these disinfected spores in PDA. Daily observation of the plates showed different structures of the fungal life cycle, as observed in Figures 1A and B where we show the formation of probasidia and basidiospores. After 10 days, 0.72±0.01% teliospores germinated, leading to the development of typical T. frezii mycelium. Nuclei stained with propidium iodide and observed in fluorescence microscopy revealed that 100% of the stained hyphae were monokaryotic (Fig. 1C). Under these conditions, 200 genes were differentially expressed: 134 were up-regulated, and 66 were down-regulated in the hyphal stage. Based on the RNA sequences obtained, protein sequences were deduced and analyzed by BLASTp of non-redundant protein sequences in the National Center for Biotechnology Information (NCBI)36.
 Germination and hyphae growth from teliospore after 5 days of incubation. (B) Formation of mycelium after 10 days of incubation. (C) Nuclear stain of hyphae with propidium iodide. This dye binds to the genetic material and stains it in red. The addition of RNAse in the staining process removes the RNA allowing the visualization of the nucleus only.")
Germination of teliospores in PDA plates. (A) Germination and hyphae growth from teliospore after 5 days of incubation. (B) Formation of mycelium after 10 days of incubation. (C) Nuclear stain of hyphae with propidium iodide. This dye binds to the genetic material and stains it in red. The addition of RNAse in the staining process removes the RNA allowing the visualization of the nucleus only.
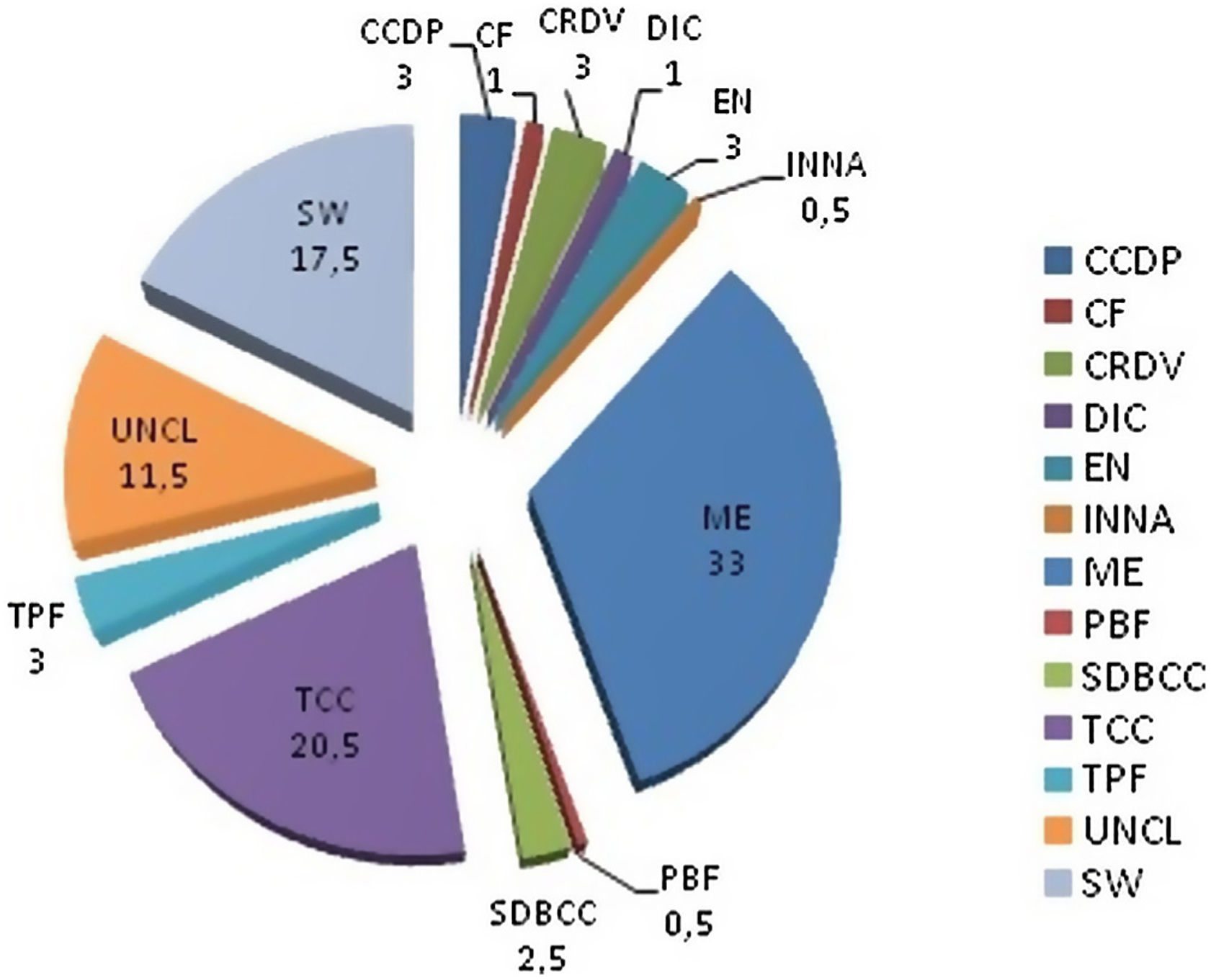
Figure 2 displays the functional grouping of all the genes that exhibited differential expression in response to the germination of teliospores into hyphae. We classified those genes into: (1) cell cycle and DNA processing (CCDP); (2) cell fate (CF); (3) cell rescue, defense and virulence (CRDV); (4) detoxification involving CYP450 (DIC); (5) energy (EN); (6) interaction of nutrients and nutritional adaptation (INNA); (7) metabolism (ME); (8) protein with binding function or cofactor requirement (PBF); (9) stress, cell differentiation and biogenesis of cellular components (SDBCC); (10) transport and cellular communication (TCC); (11) transcription (TPF); (12) unclassified (UNCL) proteins and (13) involved in several metabolic pathways (SW).
. (1) Cell cycle and DNA processing (CCDP); (2) cell fate (CF); (3) cell rescue, defense and virulence (CRDV); (4) detoxification involving CYP450 (DIC); (5) energy (EN); (6) interaction of nutrients and nutritional adaptation (INNA); (7) metabolism (ME); (8) protein with binding function or cofactor requirement (PBF); (9) stress, cell differentiation and biogenesis of cellular components (SDBCC); (10) transport and cellular communication (TCC); (11) transcription (TPF); (12) unclassified (UNCL) proteins and (13) implicated in several metabolic pathways (SW).")
Functional gene categorization (%). (1) Cell cycle and DNA processing (CCDP); (2) cell fate (CF); (3) cell rescue, defense and virulence (CRDV); (4) detoxification involving CYP450 (DIC); (5) energy (EN); (6) interaction of nutrients and nutritional adaptation (INNA); (7) metabolism (ME); (8) protein with binding function or cofactor requirement (PBF); (9) stress, cell differentiation and biogenesis of cellular components (SDBCC); (10) transport and cellular communication (TCC); (11) transcription (TPF); (12) unclassified (UNCL) proteins and (13) implicated in several metabolic pathways (SW).
From the total number of differentially expressed genes, three categories showed the greatest contribution: those involved in metabolism (Table 1; 66 genes); in transport and cellular communication (Table 2; 41 genes) and those encoding unclassified proteins (Table S2; 23 genes), with a contribution of 33%, 20.5% and 11.5% respectively.
Genes involved in metabolism.
| GenBank accession number | Gene description | Fold changeT→H (times) |
|---|---|---|
| MW691228 | Uricase | 92.50 UP |
| MW691229 | Haloacid dehalogenase-like hydrolase | 85.75 UP |
| MW691230 | Putative dihydroxy-acid dehydratase | 69.27 UP |
| MW691231 | Catalase/peroxidase HPI | 60.44 UP |
| MW691232 | Related to ADH6 – NADPH-dependent alcohol dehydrogenase | 51.26 UP |
| MW691233 | Putative formate dehydrogenase (NAD+) | 32.16 UP |
| MW691234 | Putative phosphomannomutase | 31.51 UP |
| MW691235 | Putative sterol C-24 reductase | 23.02 UP |
| MW691236 | Glutaminase A | 19.74 UP |
| MW691237 | K homology RNA-binding domain | 17.49 UP |
| MW691238 | Putative adenylosuccinate synthase | 15.40 UP |
| MW691239 | Cytochrome-c peroxidase | 14.08 UP |
| MW691240 | Acetyl-CoA synthetase | 14.03 UP |
| MW691241 | Related to deacetylase | 13.51 UP |
| MW691242 | Related to b2-aldehyde-forming enzyme | 13.11 UP |
| MW691243 | Glycosyltransferase family 1 | 11.28 UP |
| MW691244 | Putative alanine/arginine aminopeptidase | 11.09 UP |
| MW691245 | Short chain dehydrogenase | 10.75 UP |
| MW691246 | Putative glycine decarboxylase subunit P | 10.27 UP |
| MW691247 | Putative inositol polyphosphate kinase | 9.95 UP |
| MW691248 | Glycosyltransferase family 20 | 9.02 UP |
| MW691249 | PTPLA-domain-containing protein | 9.00 UP |
| MW691250 | Nucleotide-binding domain of the sugar kinase/HSP70/actin | 8.84 UP |
| MW691251 | FBP1 (fructose-1,6-bisphosphatase 1) | 8.76 UP |
| MW691252 | Putative bifunctional (2E,6E)-farnesyl diphosphate synthase/dimethylallyltranstransferase | 8.75 UP |
| MW691253 | Glyceraldehyde 3-phosphate dehydrogenase | 8.40 UP |
| MW691254 | Putative bifunctional purine biosynthetic protein ade1 | 8.16 UP |
| MW691255 | Asparagine synthase | 7.99 UP |
| MW691256 | CoA-transferase family III | 7.88 UP |
| MW691257 | Putative adenylyl-sulfate kinase | 7.72 UP |
| MW691258 | Putative pre-mRNA splicing factor prp1 | 7.52 UP |
| MW691259 | Putative histidine biosynthesis trifunctional protein | 7.42 UP |
| MW691260 | Putative xylulokinase | 7.31 UP |
| MW691261 | 2-Methylcitrate dehydratase | 7.12 UP |
| MW691262 | Aspartate aminotransferase | 6.78 UP |
| MW691263 | Putative triose-phosphate isomerase TPI1 | 6.29 UP |
| MW691264 | Putative molybdenum cofactor synthesis protein | 6.27 UP |
| MW691265 | 3-Hydroxy-3-methylglutaryl-CoA lyase | 6.21 UP |
| MW691266 | 5-Formyltetrahydrofolate cyclo-ligase family | 6.06 UP |
| MW691267 | Putative 5-methyltetrahydropteroyltriglutamate-homocysteine S-methyltransferase | 6.06 UP |
| MW691268 | Glycerol 2-dehydrogenase (NADP(+)) | 4.87 UP |
| MW691269 | Probable UDP-galactopyranose mutase | 4.87 UP |
| MW691270 | UTP-glucose-1-phosphate uridyltransferase | 4.72 UP |
| MW691271 | Putative fumarate reductase | 4.48 UP |
| MW691272 | Probable LYS12 (homo-isocitrate dehydrogenase) | 4.43 UP |
| MW691273 | Putative aspartate aminotransferase | 4.42 UP |
| MW691274 | Conserved hypothetical protein | 3.41 UP |
| MW691275 | GLN1 | 3.08 UP |
| MW691276 | HOM6 | 1.57 UP |
| MW691277 | Related to betaine lipid synthase | 1.37 UP |
| MW691278 | Uncharacterized protein | 1.31 UP |
| MW691279 | ILV5-ketol-acid reductoisomerase | 1.27 UP |
| MW691280 | Uncharacterized protein | 1.04 DOWN |
| MW691281 | Fatty acid elongase | 1.56 DOWN |
| MW691282 | Aldo-keto reductase yakc [NADP+] | 1.63 DOWN |
| MW691283 | Fer6 – related to ATP-binding cassette transporter protein | 1.91 DOWN |
| MW691284 | Fer5 – related to N6-hydroxylysine acetyltransferase | 2.74 DOWN |
| MW691285 | 5′-AMP-activated protein kinase catalytic subunit alpha-2 | 5.21 DOWN |
| MW691286 | Peptide methionine sulfoxide reductase | 6.62 DOWN |
| MW691287 | Inositol phospholipid synthesis and fat-storage-inducing TM | 8.56 DOWN |
| MW691288 | Related to 7alpha-cephem-methoxylase P8 chain | 8.33 DOWN |
| MW691289 | Putative carboxyl methyl esterase | 10.48 DOWN |
| MW691290 | UDP-glucose 6-dehydrogenase | 11.96 DOWN |
| MW691291 | Sodium-independent sulfate anion transporter | 16.30 DOWN |
| MW691292 | Related to peroxisomal amine oxidase (copper-containing) | 18.86 DOWN |
| MW691293 | Pyridoxal 5′-phosphate synthase lyase subunit PdxS | 20.21 DOWN |
T: teliospore; H: hypha.
Genes involved in transport and cellular communication.
| GenBank accession number | Gene description | Fold change T T→H (times) |
|---|---|---|
| MW691294 | Mitogen-activated serine/threonine-protein kinase | 65.56 UP |
| MW691295 | Related to aquaporin | 25.79 UP |
| MW691296 | Putative monosaccharide transporter | 17.53 UP |
| MW691297 | Putative aldehyde dehydrogenase family 7 member A1 | 16.45 UP |
| MW691298 | Putative nicotinate-nucleotide diphosphorylase (carboxylation) | 16.08 UP |
| MW691299 | Probable MET22 – protein ser/thr phosphatase | 13.65 UP |
| MW691300 | Sugar transport protein | 10.66 UP |
| MW691301 | Aspartic protease | 9.34 UP |
| MW691302 | Cation efflux family | 8.28 UP |
| MW691303 | Mitochondrial carrier protein | 7.84 UP |
| MW691304 | Mitochondrial carrier protein | 7.82 UP |
| MW691305 | Transmembrane amino acid transporter protein | 7.36 UP |
| MW691306 | Putative heat shock protein Hsp88 | 7.24 UP |
| MW691307 | Mitochondrial matrix Mmp37 | 6.46 UP |
| MW691308 | Conserved hypothetical ATP-binding protein | 6.34 UP |
| MW691309 | Hexose transporter | 6.29 UP |
| MW691310 | Major facilitator superfamily | 6.27 UP |
| MW691311 | ABC transporter | 3.25 UP |
| MW691312 | Uncharacterized protein | 2.37 UP |
| MW691313 | Rac1 GTP-binding protein | 2.00 UP |
| MW691314 | Related to putative monooxygenase | 1.13 UP |
| MW691315 | Related to acetate kinase | 1.59 DOWN |
| MW691316 | Related to MDR1 – Mac1p interacting protein | 1.55 DOWN |
| MW691317 | Probable monosaccharide transporter | 1.70 DOWN |
| MW691318 | Uncharacterized protein | 3.80 DOWN |
| MW691319 | Choline transporter-like protein 2 | 4.22 DOWN |
| MW691320 | Autophagy-related protein 8 | 4.37 DOWN |
| MW691321 | Related to opsin-1 | 4.96 DOWN |
| MW691322 | Uncharacterized protein | 5.39 DOWN |
| MW691323 | SNF7 family protein | 5.88 DOWN |
| MW691324 | ESCRT-III subunit protein SNF7 | 7.10 DOWN |
| MW691325 | RasGEF domain | 7.47 DOWN |
| MW691326 | Derlin-2 | 7.49 DOWN |
| MW691327 | Putative ATP-binding cassette glutathione S-conjugate transporter | 7.80 DOWN |
| MW691328 | Putative GTP-binding protein ypt5 | 8.36 DOWN |
| MW691329 | Putative vacuolar ATP synthase subunit D | 8.99 DOWN |
| MW691330 | Putative iron-Sulfur cluster nifU-like protein | 16.20 DOWN |
| MW691331 | Lysine-specific permease | 17.02 DOWN |
| MW691332 | Plasma membrane P-type ATPase | 19.67 DOWN |
| MW691333 | Amino acid permease | 35.90 DOWN |
| MW691334 | Probable ZRT2 – zinc transporter II | 37.81 DOWN |
T: teliospore; H: hypha.
When we analyzed genes expressed and involved in the metabolism category (Table 1), 52 up-regulated and 14 down-regulated genes were found in T. frezii growth from teliospores to hyphae. Among up-regulated genes in teliospores, we found the cytochrome-C peroxidase gene (MW691239) transcript increased 14.08 times in teliospores, and GAPDH transcript (MW691253), its level is 8.40-fold higher in the teliospore stage, compared to the hyphal stage. In the same group, we observed an 85.75-fold increase in haloacid dehalogenase-like hydrolase (MW691229) in T. frezii teliospores, and a 19.74-fold increase was measured in this stage for glutaminase A enzyme (MW691236). The enzyme 2-methylcitrate dehydratase (MW691261) was observed in T. frezii teliospores, the transcription level experienced a 7.12-fold increase, compared to hyphae. The gene glycerol 2-dehydrogenase (NADP(+)) (MW691268), which plays a role in glycerol catabolism, exhibited a substantial 4.87-fold increase in expression levels within T. frezii teliospores compared to hyphae. Similarly, the RNA expression of UTP-glucose-1-phosphate uridyltransferase (MW691270) was enhanced by 4.72-fold in teliospores of T. frezii.
Within genes down-regulated in teliospores, we found that the aldo-keto reductase yakc [NADP+] (MW691282), an enzyme from the reductase superfamily, exhibited a 1.63-fold decrease in T. frezii teliospores in relation to hyphae. Additionally, we observed down-regulation in two other gene expressions: Fer6, associated with ATP-binding cassette transporter protein (MW691283), showed a 1.91-fold decrease, while Fer5, linked to N6-hydroxylysine acetyl transferase (MW691284), exhibited a 2.74-fold decrease. A decrease in the expression of two additional genes was observed in teliospores of T. frezii. The gene related to 7 alpha-cephem-methoxylase P8 chain (MW691288) showed a significant 8.33-fold decrease, while the expression of the gene related to peroxisomal amine oxidase (copper-containing) (MW691292) was down-regulated by a substantial 18.86-fold.
In the TCC category (Table 2), we observed a total of 21 up-regulated genes and 20 down-regulated genes in T. frezii during the transition from teliospores to hyphae. Among the up-regulated genes, we identified the aspartic protease (MW691301) gene, which exhibited a substantial 9.34-fold increase in teliospore expression. Additionally, the genes probable MET22 – protein ser/thr phosphatase (MW691299) and related to putative monooxygenase (MW691314) were also found to be up-regulated in T. frezii teliospores, with fold increases of 13.65 and 1.13, respectively.
One of the most prominently down-regulated genes was the amino acid permease (MW691333), which exhibited a substantial 35.90-fold decrease in expression in T. frezii teliospores. Additionally, among the other down-regulated genes, we observed a decrease in expression for those related to acetate kinase (MW691315), to opsin-1 (MW691321), and an uncharacterized protein (um02763) (MW691322) with fold decreases of 1.59, 4.96, and 5.39, respectively. Furthermore, in relation to the autophagy-related protein (MoAtg14) (MW691320), T. frezii showed a significant 4.37-fold decrease in expression levels when comparing teliospores to hyphae.
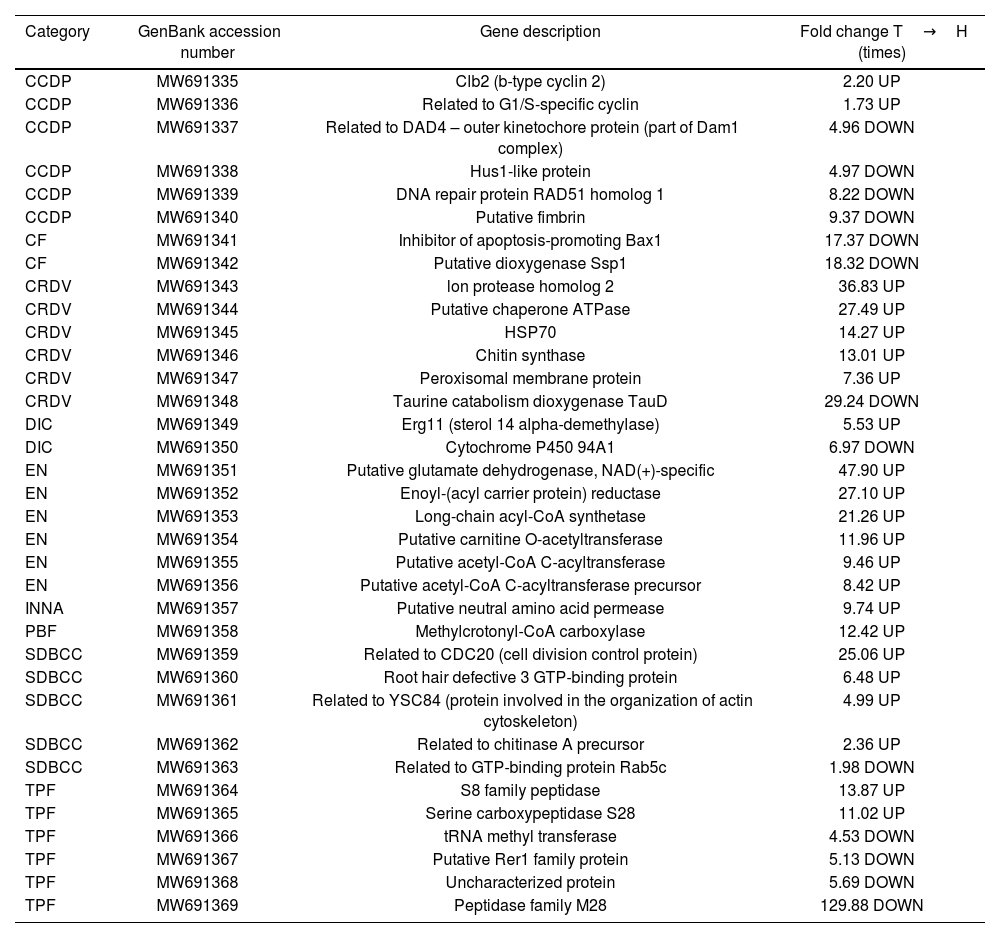
In the categories of CCDP, CF, CRDV, DIC, EN, INNA, PBF, SDBCC and TPF (Table 3), 22 up-regulated genes were found (2, 0, 5, 1, 6, 1, 1, 4 and 2 respectively) and 13 down-regulated genes (4, 2, 1, 1, 0, 0, 0, 1 and 4 respectively) in T. frezii transition from teliospores to hyphae.
Genes involved in CCDP, CF, CRDV, DIC, EN, INNA, PBF, SDBCC and TPF.
| Category | GenBank accession number | Gene description | Fold change T→H (times) |
|---|---|---|---|
| CCDP | MW691335 | Clb2 (b-type cyclin 2) | 2.20 UP |
| CCDP | MW691336 | Related to G1/S-specific cyclin | 1.73 UP |
| CCDP | MW691337 | Related to DAD4 – outer kinetochore protein (part of Dam1 complex) | 4.96 DOWN |
| CCDP | MW691338 | Hus1-like protein | 4.97 DOWN |
| CCDP | MW691339 | DNA repair protein RAD51 homolog 1 | 8.22 DOWN |
| CCDP | MW691340 | Putative fimbrin | 9.37 DOWN |
| CF | MW691341 | Inhibitor of apoptosis-promoting Bax1 | 17.37 DOWN |
| CF | MW691342 | Putative dioxygenase Ssp1 | 18.32 DOWN |
| CRDV | MW691343 | lon protease homolog 2 | 36.83 UP |
| CRDV | MW691344 | Putative chaperone ATPase | 27.49 UP |
| CRDV | MW691345 | HSP70 | 14.27 UP |
| CRDV | MW691346 | Chitin synthase | 13.01 UP |
| CRDV | MW691347 | Peroxisomal membrane protein | 7.36 UP |
| CRDV | MW691348 | Taurine catabolism dioxygenase TauD | 29.24 DOWN |
| DIC | MW691349 | Erg11 (sterol 14 alpha-demethylase) | 5.53 UP |
| DIC | MW691350 | Cytochrome P450 94A1 | 6.97 DOWN |
| EN | MW691351 | Putative glutamate dehydrogenase, NAD(+)-specific | 47.90 UP |
| EN | MW691352 | Enoyl-(acyl carrier protein) reductase | 27.10 UP |
| EN | MW691353 | Long-chain acyl-CoA synthetase | 21.26 UP |
| EN | MW691354 | Putative carnitine O-acetyltransferase | 11.96 UP |
| EN | MW691355 | Putative acetyl-CoA C-acyltransferase | 9.46 UP |
| EN | MW691356 | Putative acetyl-CoA C-acyltransferase precursor | 8.42 UP |
| INNA | MW691357 | Putative neutral amino acid permease | 9.74 UP |
| PBF | MW691358 | Methylcrotonyl-CoA carboxylase | 12.42 UP |
| SDBCC | MW691359 | Related to CDC20 (cell division control protein) | 25.06 UP |
| SDBCC | MW691360 | Root hair defective 3 GTP-binding protein | 6.48 UP |
| SDBCC | MW691361 | Related to YSC84 (protein involved in the organization of actin cytoskeleton) | 4.99 UP |
| SDBCC | MW691362 | Related to chitinase A precursor | 2.36 UP |
| SDBCC | MW691363 | Related to GTP-binding protein Rab5c | 1.98 DOWN |
| TPF | MW691364 | S8 family peptidase | 13.87 UP |
| TPF | MW691365 | Serine carboxypeptidase S28 | 11.02 UP |
| TPF | MW691366 | tRNA methyl transferase | 4.53 DOWN |
| TPF | MW691367 | Putative Rer1 family protein | 5.13 DOWN |
| TPF | MW691368 | Uncharacterized protein | 5.69 DOWN |
| TPF | MW691369 | Peptidase family M28 | 129.88 DOWN |
CCDP: cell cycle and DNA processing; CF: cell fate; CRDV: cell rescue, defense and virulence; DIC: detoxification involving CYP450; EN: energy; INNA: interaction of nutrients and nutritional adaptation; PBF: protein with binding function or cofactor requirement; SDBCC: stress, cell differentiation and biogenesis of cellular components; TPF: transcription; T: teliospore; H: hypha.
In the cell cycle and DNA processing category, two genes were shown to be up-regulated in T. frezii teliospores, Clb2 (b-type cyclin 2) (MW691335) and the gene related to G1/S-specific cyclin (MW691336) (fold increases of 2.2 and 1.73, respectively). In the cell rescue, defense and virulence category, the peroxisomal membrane protein (MW691347) was 7.36-fold up-regulated at RNA level in T. frezii teliospores. We also found a 14.27-fold increase in the Hsp70 protein (MW691345) in teliospores. With regard to the synthesis of chitin, one of the most important components of the fungal cell wall, we found that one of the genes that codes for chitin synthase (MW691346) increased 13.01-fold in teliospores, compared to the hyphal stage in T. frezii. In DIC category, we found just one up-regulated gene, the sterol 14 alpha-demethylase (MW691349), an essential enzyme in sterol biosynthesis, which was 5.53-fold up-regulated in T. frezii teliospores. In SDBCC category, some of the up-regulated genes in T. frezii teliospores were those related to CDC20 – cell division control protein (MW691359), to YSC84 – a protein involved in the organization of actin cytoskeleton (MW691361) and to chitinase A precursor (MW691362) (25.06, 4.99, 2.36 respectively). One of the proteins related to nutritional adaptation function, the putative neutral amino acid permease (MW691357), was found to be 9.71-fold up-regulated in T. frezii teliospores. In the energy category, we found the enoyl-(acyl carrier protein) reductase (MW691352), which is the last enzyme in the fatty acid elongation cycle19, its expression exhibited a 27.10-fold increase in T. frezii teliospores.
In our analysis of down-regulated genes in teliospores, we observed significant decreases in the expression of two genes belonging to the cell fate category: Ssp1 protein kinase (MW691342) and the inhibitor of apoptosis-promoting Bax1 (MW691341)9. These genes exhibited a remarkable 18.32 and 17.37-fold reduction, respectively. The gene that encodes for putative Rer1 family protein (MW691367) was also down-regulated in teliospores.
Regarding the genes that participate in multiple pathways (Table S3), a total of 26 up-regulated genes and 9 down-regulated genes were identified. Notably, probable adenosylhomocysteinase (MW691399) exhibited a substantial 6.93-fold increase, while probable SMC2 – a chromosome segregation protein (MW691373) showed a modest 1.09-fold up-regulation. In contrast, PacC – a transcription factor (MW691404), experienced a remarkable 2.48-fold down-regulation. We also found three other up-regulated genes, TPM2 – tropomyosin isoform 2 (MW691380), mitochondrial porin (MW691376), and CipC homolog (MW691370), which were 7.40, 5.00, and 8.44-fold increased, respectively.
In the unclassified genes category (Table S2), during the transition from teliospores to the hyphal stage, a total of 13 up-regulated genes and 10 down-regulated genes were observed. Notably, the um10414 protein (MW691406) exhibited a significant up-regulation, with a remarkable 49.29-fold increase in gene expression. On the other hand, the genes encoding for um05276 (MW691418), um10868 (MW691421), um04575 (MW691424), and um10208 (MW691425) showed down-regulation, with their expression levels exhibiting 1.45, 1.72, 12.50, and 15.74-fold decreases, respectively.
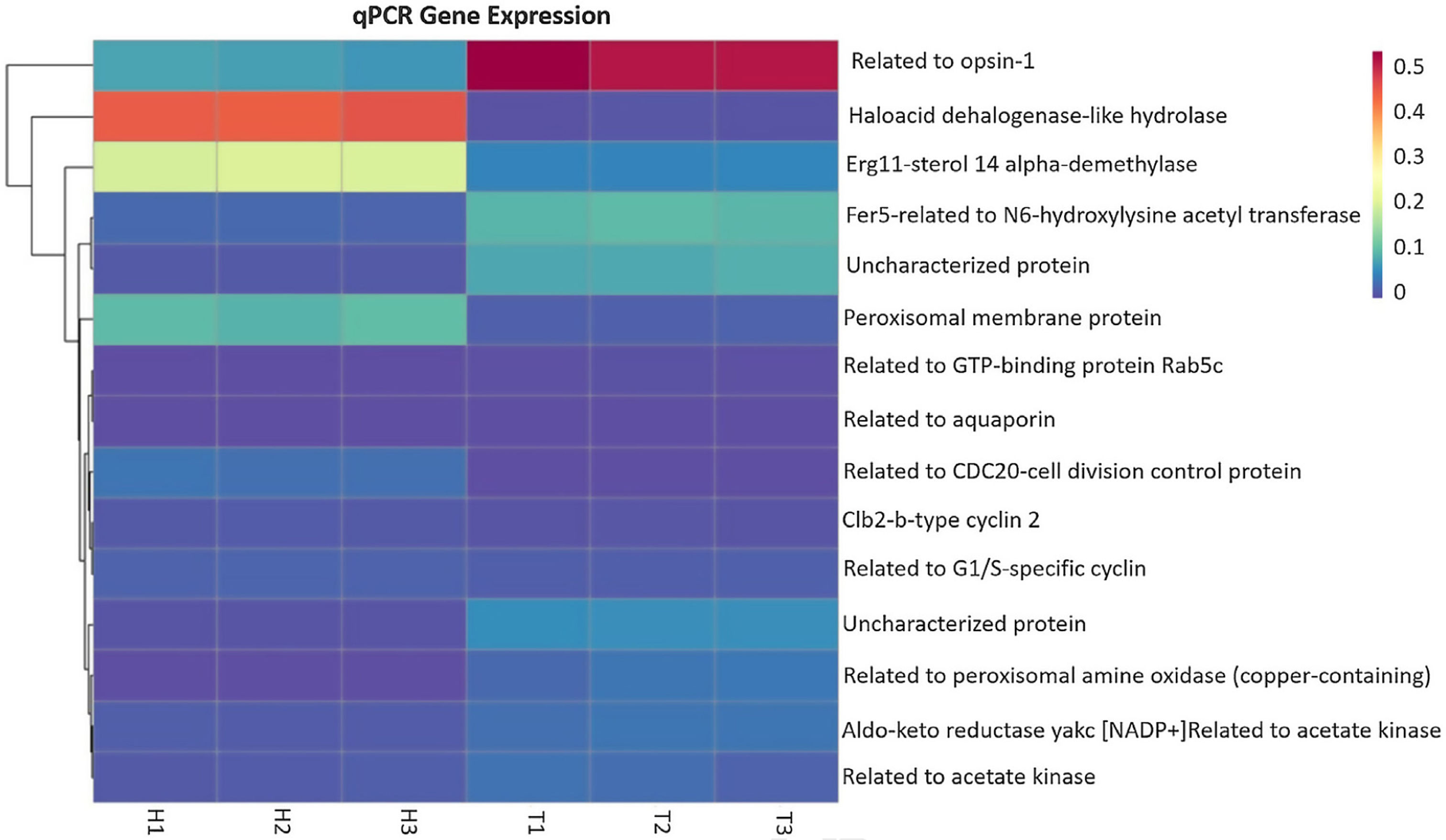
qPCR analysis of selected genesTo validate the reliability of the transcriptome sequencing, we selected 15 differentially expressed genes of the two T. frezii stages (teliospore and mycelium) for RT-qPCR analysis. The results showed that 7 of the 15 genes selected were up-regulated and 8 were down-regulated (Fig. 3). Our results were consistent with the transcriptome data. Differences were significant in all cases (p<0.02) except for those related to acetate kinase gene.
 and teliospores (T1, T2, and T3 replicates). Red and blue shadings represent higher and lower relative expression levels, respectively. Genes have been grouped based on their pattern of gene expression. Actin transcript was used as a reference gene for expression normalization.")
Expression of some differentially expressed genes quantified by real-time PCR. Heatmap is showing differential gene expression between hyphae (H1, H2, and H3 replicates) and teliospores (T1, T2, and T3 replicates). Red and blue shadings represent higher and lower relative expression levels, respectively. Genes have been grouped based on their pattern of gene expression. Actin transcript was used as a reference gene for expression normalization.
To date, no comprehensive investigation has been carried out to analyze the gene regulation occurring during the life cycle of T. frezii. Our work allowed to identify genes that are differentially expressed in two stages, teliospore and hypha. As described for other smut fungi, after teliospore germination and meiosis, haploid sporidia are formed and cell fusion of two haploid mating types leads to the development of infectious dikaryotic hyphae. This process occurs on the host plant, during the mating process between cells of opposite mating types35. However, as observed in Cryptococcus neoformans34, a shift from teliospores to monokaryotic hyphae was found in in vitroT. frezii cultures.
Genes that code for proteins involved in metabolismBy examining the expression patterns of specific genes, we identified the cytochrome-C peroxidase gene (MW691239) where the transcript showed a remarkable 14-fold increase in teliospores. It is noteworthy that in the fungus Candida albicans, this protein has been targeted for antifungal treatment using quinonemethide27, which effectively reduces its expression. Hence, considering this relationship, it could be proposed as a potential alternative for controlling T. frezii. Furthermore, our analysis of the expression levels of the GAPDH transcript (MW691253) revealed an 8-fold increase in the teliospore stage; this finding aligns with observations made in Penicillium marneffei11 where there was an impressive 20-fold increase in the expression of this gene. This gene is described to encode an enzyme that plays a crucial role in an adhesion factor for conidial attachment. We also made a discovery regarding haloacid dehalogenase-like hydrolase (MW691229) which has been previously identified as an important virulence factor in Pyrenophora teres f. teres10. Notably, the expression of this gene is substantially elevated in T. frezii teliospores. In U. maydis, the transcriptomic analysis revealed a 3-fold increase, while the protein analysis indicated a 2.0–2.2-fold induction3. Conversely, the glutaminase A enzyme (MW691236) exhibits a striking 85-fold increase in RNA levels in U. maydis, compared to a near 20-fold increase in T. frezii teliospores3. Furthermore, the enzyme 2-methylcitrate dehydratase (MW691261) exhibited a 1.81-fold increase at the protein level in U. maydis, coinciding with a nearly 7-fold increase in transcription levels in T. frezii teliospores17. Glycerol 2-dehydrogenase (NADP(+)) (MW691268) has been identified as a key player in glycerol catabolism under microaerobic conditions in U. maydis. Interestingly, in T. frezii teliospores, its expression level showed a significant nearly 5-fold increase compared to hyphae. This notable increase in protein expression was also observed in U. maydis3. Additionally, in our study, we found a consistent 5-fold increase in the expression of UTP-glucose-1-phosphate uridyltransferase (MW691270) at the protein level both in U. maydis3 and T. frezii teliospores.
Aldo-keto reductase yakc [NADP+] (MW691282), nearly experienced a 2-fold decrease in T. frezii teliospores in relation to hyphae. This enzyme from the reductase superfamily, also showed a 1.97-fold decrease (at protein level) in U. maydis17. Both Fer6, which is associated with ATP-binding cassette transporter protein (MW691283), and Fer5, linked to N6-hydroxylysine acetyl transferase (MW691284), exhibited approximately a 2-fold downregulation in T. frezii teliospores compared to hyphae. In contrast, in U. maydis, the same genes demonstrated a much more pronounced down-regulation with 80.5 and 165.4-fold decrease (RNA) respectively. A decrease in the expression of those related to 7 alpha-cephem-methoxylase P8 chain (MW691288) was observed both in teliospores of T. frezii and of U. maydis (8 and 2 times, respectively)18. In the same article, the expression of those related to peroxisomal amine oxidase (copper-containing) (MW691292) was found to be down-regulated by 61-fold in U. maydis18 and we found a nearly 19-fold decrease in teliospores of T. frezii.
Genes involved in transport and cellular communicationDuring our gene expression analysis focused on proteins involved in transport and cell communication processes, we made a remarkable observation regarding the expression of aspartic protease (MW691301), known as a protein expressed during the early stages of sunflower cotyledon infection by Sclerotinia sclerotiorum21. In T. frezii, in the teliospore stage, the expression of this enzyme exhibited a significant 9.34-fold increase. Notably, in Paracoccidioides brasiliensis fungi, during the infection process, this gene was up-regulated by 9.5-fold5, suggesting its potential importance in the infection process. Comparisons in the expression levels of several genes with their orthologues of U. maydis are compiled in the work of Ruiz-Herrera et al.18 Among these comparisons, it was observed that the genes encoding for probable MET22 – protein ser/thr phosphatase (MW691299) and related to putative monooxygenase (MW691314) were up-regulated in both T. frezii and U. maydis, showing 13.65 and 1.13-fold changes in T. frezii, and 2.3 and 2.5 in U. maydis, respectively.
In terms of down-regulated genes, the authors reported that the genes related to acetate kinase (MW691315), related to opsin-1 (MW691321), and uncharacterized protein (um02763) (MW691322) exhibited down-regulation in the same direction in U. maydis. The fold changes observed were 1.59, 4.96, and 5.39 in T. frezii, and 10.9, 5, and 114 in U. maydis, respectively. One of the most significant down-regulated genes identified was the amino acid permease (MW691333), which plays a fundamental role in fluconazole resistance and is the target of plant secondary metabolites with antifungal properties. In U. maydis, it exhibited specific expression during budding6, while in T. frezii hyphae, its expression showed a 36-fold increase. With regard to the autophagy-related protein (MoAtg14) (MW691320), a study conducted by Liu et al.12, demonstrated that the deletion of this protein in Magnaporthe oryzae resulted in a complete loss of virulence and conidiation problems. However, contrary to our expectations, T. frezii, exhibited a decreased 4.37-fold expression during the fungi transition.
Genes that code for proteins involved in CCDP, CF, CRDV, DIC, EN, INNA, PBF, SDBCC and TPFIn the cell cycle and DNA processing category, only two genes were shown to be up-regulated during the transition in both T. frezii and U. maydis, Clb2 (b-type cyclin 2) (MW691335) and those related to G1/S-specific cyclin (MW691336)18.
In the cell rescue, defense, and virulence category, the peroxisomal membrane protein (MW691347) exhibited a significant up-regulation of nearly 7-fold in T. frezii. Similarly, at the protein level, it showed a 2.31-fold up-regulation in U. maydis17. Another up-regulated gene encoding for the Hsp70 protein (MW691345) is considered essential for many cellular processes and plays a major role in various stress conditions. Its synthesis is induced by heat shock stress, as observed in U. maydis with a 5-fold increase in heating conditions8. It has also been observed to be up-regulated during plant pathogenesis32. Concerning the synthesis of chitin, we found that one of the genes that codes for chitin synthase (MW691346)30 is 13-fold up-regulated in teliospores of T. frezii. Interestingly, this gene has been previously reported to be exclusive to the hyphae stage in the fungus Mucor circinelloides14, highlighting its differential expression during the transition process. These findings support the existence of a multigene chitin synthase family, where expression varies according to the fungal stage. This suggests that different chitin synthase activities may play distinct roles in the dimorphic growth of M. circinelloides.
Another up-regulated gene expressed in T. frezii is sterol 14 alpha-demethylase (MW691349), which participates in sterol biosynthesis in eukaryotes and is a clinical target for antifungal azoles, and was also up-regulated in U. maydis18 (2.7 times). The authors also reported up-regulation of genes related to CDC20 – cell division control protein (MW691359), YSC84 (MW691361) and chitinase A precursor (MW691362) in U. maydis18 (with 2.5, 2.0, 6.7-fold increases, respectively). A notable virulence factor found in various fungi, including Saccharomyces cerevisiae and Candida albicans, is associated with a neutral amino acid permease (MW691357)16. Research reveals that these fungi possess twenty-four and twenty-seven genes, respectively, that encode for this protein, further contributing to their virulence.
The enoyl-(acyl carrier protein) reductase (ENRs), an enzyme that belongs to the energy category, was remarkably up-regulated in teliospores. This is interesting because this protein catalyzes the last step of the elongation cycle in the synthesis of fatty acids, whose biosynthesis is essential for survival in mammals, plants, fungi and bacteria19. The growing interest in ENRs is mainly due to the fact that a variety of antimicrobial compounds, such as triclosan, are shown to specifically target their activity.
The Ssp1 protein kinase (MW691342), identified as one of the down-regulated genes in the CF category, plays a crucial role in facilitating the reorganization of the actin cytoskeleton following osmotic stress. In T. frezii, its expression increased by 18-fold in the hyphal stage compared to the teliospore stage. However, in U. maydis, during the transition from the budding to the filamentous stage, this increase was comparatively lower, at only 4-fold6. Putative Rer1 family protein (MW691367) is involved in the retrieval of endoplasmic reticulum membrane proteins from the early Golgi compartment. In T. frezii, this gene expression was 5.13-fold decreased in teliospores, while in M. haptotylum knobs/mycelium transition, the down-expression was 1.361.
Martínez-Soto et al.18, found many genes differentially expressed in the transition between spore and hyphae in U. maydis, particularly, three genes, two of which were up-regulated and one was down-regulated: probable adenosylhomocysteinase (MW691399), probable SMC2 – chromosome segregation protein (MW691373) and pacC – transcription factor pacC (MW691404) (2 (up); 10.3 (up) and 9.2 (down) fold, respectively). T. frezii orthologues genes were also differentially expressed and in the same direction (up- or down-regulated). Finally, three other genes were up-regulated in the U. maydis dimorphism17, TPM2 (tropomyosin isoform 2 (MW691380), a component of fungal filaments), mitochondrial porin (MW691376) (which allows the passage of small molecules across the mitochondrial outer membrane and is involved in complex interactions regulating organelle and cellular metabolism) and CipC homolog (MW691370) (5.95; 2.25 and 14.04-fold at protein level, respectively). Coincidentally, we observed that the orthologues of these genes in T. frezii were also up-regulated.
At least five uncharacterized proteins were differentially expressed in both T. frezii and U. maydis. In the transition of U. maydis, the variation of the transcripts of the um10414 protein was 3.5-fold increased; a trend that was also observed in its orthologous protein in T. frezii. In contrast, four other proteins, um05276, um10868, um04575 and um10208 displayed down-regulation with fold changes of 11.7; 2.7; 2.6 and 4.7-fold decrease, respectively. Similarly, the transcripts levels of their orthologous proteins in T. frezii (MW691418, MW691421, MW691424 and MW691425) were also down-regulated.
In conclusion, many cellular processes are involved in the morphological shifts from teliospores to hyphae of T. frezii, which makes it difficult to fully understand the intricacies of this complex process. For this reason, different approaches are necessary before we can explain this process at a molecular level. Consequently, we believe that our comprehensive analysis of the entire transcriptome during this transition serves as a fundamental step toward understanding this phenomenon and may lay the foundation for future studies of the molecular processes that occur during infection.
Conflicts of interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
The present work was supported by grants from Universidad Católica de Córdoba, Centro de Excelencia en Productos y Procesos de Córdoba (CEPROCOR), Fundación Maní Argentino and Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET).
The following are the supplementary data to this article:
