





Brucella abortus es el agente causal de la brucelosis bovina, enfermedad zoonótica que se encuentra ampliamente distribuida en el mundo. Actualmente existen ocho biovariedades de B. abortus. En Argentina se encuentra con mayor frecuencia la biovariedad 1, pero también se suele aislar la biovariedad 2, que es más patogénica que la anterior. Resulta necesario contar con métodos de tipificación que tengan la resolución suficiente para permitir el seguimiento epidemiológico de los brotes de brucelosis y de los programas de control de la enfermedad. Debido a la gran homogeneidad genética que existe entre las distintas especies del género Brucella, ha sido dificultoso el desarrollo de herramientas moleculares para realizar el análisis epidemiológico de los aislamientos. La publicación del genoma de varias especies de Brucella facilitó el diseño de estas herramientas. El objetivo del presente trabajo fue emplear un esquema de análisis multilocus de VNTR en aislamientos de Argentina obtenidos en nuestro laboratorio. De los 56 aislamientos analizados se obtuvieron 47 perfiles genotípicos diferentes. El empleo de este esquema permitió asignarles a dichos aislamientos la biovariedad correspondiente. A través del análisis goeBURST se pudo relacionar a todos los genotipos entre sí, y además, proponer al genotipo de la biovariedad 2 como fundador.
Brucella abortus is the causative agent of bovine brucellosis, a worldwide zoonosis. Up to date, eight biovars of B. abortus have been described. In Argentina, biovar 1 is the most frequently isolated. However, biovar 2, which is more pathogenic than biovar 1, is also found. Molecular methods for subtyping isolates are necessary for allowing epidemiological surveillance and control of eradication programs. Due to the genetic homogeneity of the genus Brucella, the development of molecular typing tools has been difficult. The publication of microorganism genomes facilitates the design of this approach. The aim of this work was to employ a Multiple Locus VNTR Analysis (MLVA) scheme for strains from Argentina isolated in our laboratory. From the 56 isolates analyzed, 47 different genotypic profiles were obtained. All the strains typed as biovar 2 showed the same profile. This scheme allowed assigning each isolate to the biovar it belongs to. All the genotypes were related using the goeBURST analysis and biovar 2 was proposed as founder.
La brucelosis es una zoonosis que se encuentra principalmente en América Latina, en la región del Mediterráneo en Europa, en el oeste de Asia y en gran parte del territorio africano. Esta enfermedad está causada por bacterias del género Brucella, microorganismo gram negativo, intracelular facultativo. Se reconocen varias especies dentro del género, siendo Brucella abortus la causante de la brucelosis bovina.
Existen ocho biovariedades descritas de B. abortus1. En Argentina se encuentra con mayor frecuencia la biovariedad 1, pero también se suele aislar de animales y humanos13 la biovariedad 219. Esta biovariedad se ha observado en infecciones naturales en bovinos, asociada a la «tormenta de abortos» que provoca en los animales preñados infectados, y al nacimiento de terneros que mueren a las horas y también de «natimortos», es decir, de terneros que nacen en término pero muertos. En infecciones experimentales en cobayos, B. abortus biovariedad 2 produjo lesiones granulomatosas mucho más graves que la biovariedad 1 (Samartino L, comunicación personal). Además del fuerte impacto en la salud pública, la infección por Brucella genera grandes pérdidas económicas20.
Es sumamente útil contar con métodos de tipificación que tengan la resolución suficiente para permitir un seguimiento epidemiológico de los brotes de brucelosis y de los programas de control de la enfermedad. El desarrollo de herramientas moleculares para identificar y tipificar aislamientos del género Brucella ha sido muy laborioso debido a la falta de polimorfismo dentro del género. Las especies de Brucella presentan una homología intraespecífica mayor del 80 % por estudios de hibridación ADN-ADN23, una similitud de secuencia mayor del 98 % por genómica comparativa9, y la secuenciación del ARNr 16S muestra un 100 % de identidad entre todas las especies de Brucella7.
Dada la homogeneidad genética que se observa dentro del género Brucella, es de esperar que marcadores basados en secuencias génicas no provean la resolución requerida para hacer un seguimiento epidemiológico. La presencia de unidades repetidas en tándem, que varían en su número dentro de regiones no codificantes del genoma, surgen de errores durante la replicación12 o de eventos de recombinación desiguales18. Como consecuencia, estos lugares puntuales presentan una alta tasa de mutación, lo que los hace muy útiles a la hora de buscar polimorfismos que permitan tipificar especies bacterianas con genomas muy conservados, como Bacillus anthracis10 y varias especies de micobacterias15.
La secuenciación de los genomas facilitó el desarrollo de nuevos métodos para tipificar Brucella, como tipificación multilocus de secuencias [Multi Locus Sequence Typing (MLST)] y análisis multilocus de repeticiones en tándem de número variable [Multiple Locus Variable Number Tandem Repeats (VNTR) Analysis (MLVA)].
El diseño racional de un esquema de tipificación mediante MLVA debería incluir marcadores con diferentes niveles de diversidad y poder discriminatorio. Así, se podrían agrupar los marcadores en dos conjuntos: uno compuesto por marcadores muy variables, que permitan diferenciar a las distintas cepas, y otro formado por marcadores más estables, con baja diversidad, menos discriminativos, para diferenciar a nivel especie. Desde una perspectiva práctica, los primeros resultarían muy útiles a la hora de realizar un estudio epidemiológico en un brote puntual de la enfermedad, mientras que los segundos permitirían relacionar aislamientos en una escala regional o global. Ejemplos de este tipo de diseño se pueden encontrar en los trabajos de Whatmore et al.24 y Le Flèche et al.11.
La utilidad de los MLVA en Brucella está bien documentada. García-Yoldi et al.6 compararon MLVA con otros métodos basados en PCR para tipificar B. suis. Los resultados obtenidos con las distintas metodologías fueron concordantes y además, los MLVA permitieron realizar inferencias sobre la relación epidemiológica de las cepas. Empleando el mismo esquema de 10 loci VNTR, Beja-Pereira et al.2 sugirieron que un brote de brucelosis bovina en el área del Parque Yellowstone (Estados Unidos) se originó a partir de renos (Cervus elaphus) y no de bisones (Bos bison), como se pensaba hasta ese momento. Higgins et al.8, por su parte, realizaron un estudio epidemiológico de B. abortus aislada de ganado (Bos primigenius), renos y bisones; esto les permitió tomar decisiones sobre el manejo de estas tres especies que conviven en la misma área. Lucero et al.14 describieron dos brotes de brucelosis humana por consumo de queso sin pasteurizar en Argentina (una familia de San Juan y otra de Buenos Aires), y mediante MLVA determinaron que los aislamientos obtenidos a partir de las personas infectadas de cada familia constituyen dos clusters genéticos diferentes.
Bricker et al.3 describieron un esquema de MLVA que llamaron HOOF-Prints, basado en la repetición en tándem de una secuencia de 8 nucleótidos que se encuentra en ocho regiones del genoma. En un trabajo posterior, Bricker et al.4 evaluaron el desempeño de los HOOF-Prints en cepas de B. abortus aisladas en Estados Unidos a través del análisis de características del método como tipabilidad, reproducibilidad, estabilidad, poder discriminatorio y concordancia epidemiológica y los compararon con otros métodos de tipificación obteniendo resultados que superan el valor mínimo sugerido por el Grupo Europeo de Estudio de Marcadores Epidemiológicos [European Study Group on Epidemiological Marker (ESGEM)]21. En 2009, Valdezate et al.22 reportaron el surgimiento de un linaje clonal de B. abortus biovariedad 3 en aislamientos clínicos en España, los HOOF-Prints les permitieron revelar la relación genética entre dichos aislamientos.
El objetivo de este trabajo fue genotipificar aislamientos de B. abortus en Argentina utilizando un esquema de MLVA combinado, el cual incluye los HOOF-Prints y algunos de los loci VNTR descritos por Whatmore et al.24.
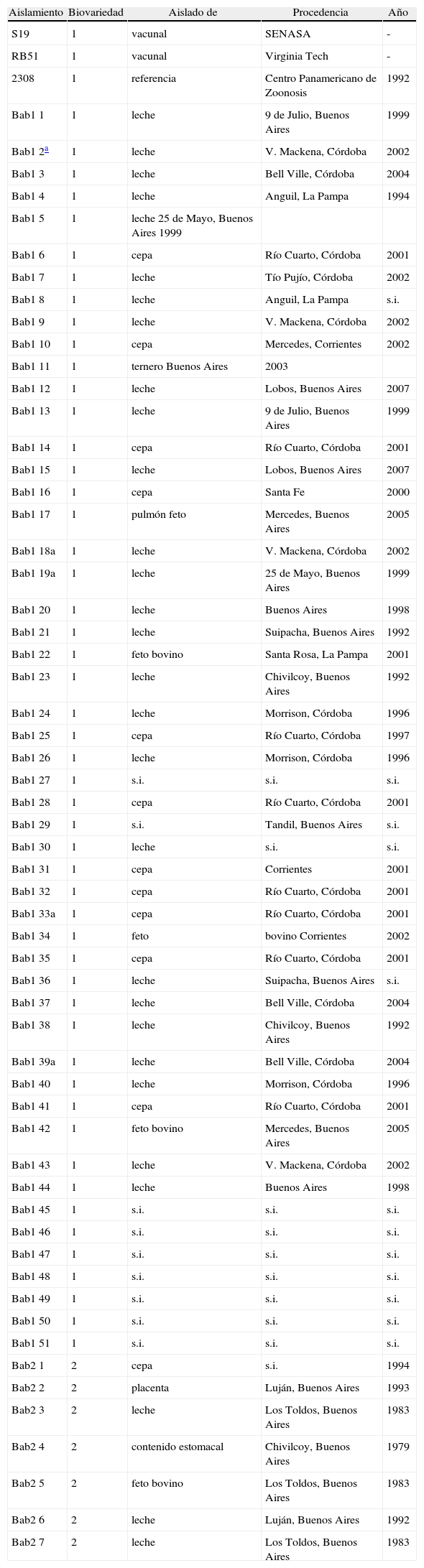
Materiales y métodosAislamientos, tipificación bioquímica y preparación del templado de PCRSe utilizaron las cepas de B. abortus conservadas en el cepario del Laboratorio de Brucelosis del Instituto de Patobiología del CICVyA de INTA. De las cepas incluidas en este estudio, 37 fueron aisladas por personal del laboratorio a partir de muestras de origen bovino, 12 fueron enviadas para su tipificación, en tanto que se desconoce la procedencia de 9 de ellas. Las cepas utilizadas en este trabajo se presentan en la tabla 1.
Características de las cepas utilizadas en este estudio.
| Aislamiento | Biovariedad | Aislado de | Procedencia | Año |
| S19 | 1 | vacunal | SENASA | - |
| RB51 | 1 | vacunal | Virginia Tech | - |
| 2308 | 1 | referencia | Centro Panamericano de Zoonosis | 1992 |
| Bab1 1 | 1 | leche | 9 de Julio, Buenos Aires | 1999 |
| Bab1 2a | 1 | leche | V. Mackena, Córdoba | 2002 |
| Bab1 3 | 1 | leche | Bell Ville, Córdoba | 2004 |
| Bab1 4 | 1 | leche | Anguil, La Pampa | 1994 |
| Bab1 5 | 1 | leche 25 de Mayo, Buenos Aires 1999 | ||
| Bab1 6 | 1 | cepa | Río Cuarto, Córdoba | 2001 |
| Bab1 7 | 1 | leche | Tío Pujío, Córdoba | 2002 |
| Bab1 8 | 1 | leche | Anguil, La Pampa | s.i. |
| Bab1 9 | 1 | leche | V. Mackena, Córdoba | 2002 |
| Bab1 10 | 1 | cepa | Mercedes, Corrientes | 2002 |
| Bab1 11 | 1 | ternero Buenos Aires | 2003 | |
| Bab1 12 | 1 | leche | Lobos, Buenos Aires | 2007 |
| Bab1 13 | 1 | leche | 9 de Julio, Buenos Aires | 1999 |
| Bab1 14 | 1 | cepa | Río Cuarto, Córdoba | 2001 |
| Bab1 15 | 1 | leche | Lobos, Buenos Aires | 2007 |
| Bab1 16 | 1 | cepa | Santa Fe | 2000 |
| Bab1 17 | 1 | pulmón feto | Mercedes, Buenos Aires | 2005 |
| Bab1 18a | 1 | leche | V. Mackena, Córdoba | 2002 |
| Bab1 19a | 1 | leche | 25 de Mayo, Buenos Aires | 1999 |
| Bab1 20 | 1 | leche | Buenos Aires | 1998 |
| Bab1 21 | 1 | leche | Suipacha, Buenos Aires | 1992 |
| Bab1 22 | 1 | feto bovino | Santa Rosa, La Pampa | 2001 |
| Bab1 23 | 1 | leche | Chivilcoy, Buenos Aires | 1992 |
| Bab1 24 | 1 | leche | Morrison, Córdoba | 1996 |
| Bab1 25 | 1 | cepa | Río Cuarto, Córdoba | 1997 |
| Bab1 26 | 1 | leche | Morrison, Córdoba | 1996 |
| Bab1 27 | 1 | s.i. | s.i. | s.i. |
| Bab1 28 | 1 | cepa | Río Cuarto, Córdoba | 2001 |
| Bab1 29 | 1 | s.i. | Tandil, Buenos Aires | s.i. |
| Bab1 30 | 1 | leche | s.i. | s.i. |
| Bab1 31 | 1 | cepa | Corrientes | 2001 |
| Bab1 32 | 1 | cepa | Río Cuarto, Córdoba | 2001 |
| Bab1 33a | 1 | cepa | Río Cuarto, Córdoba | 2001 |
| Bab1 34 | 1 | feto | bovino Corrientes | 2002 |
| Bab1 35 | 1 | cepa | Río Cuarto, Córdoba | 2001 |
| Bab1 36 | 1 | leche | Suipacha, Buenos Aires | s.i. |
| Bab1 37 | 1 | leche | Bell Ville, Córdoba | 2004 |
| Bab1 38 | 1 | leche | Chivilcoy, Buenos Aires | 1992 |
| Bab1 39a | 1 | leche | Bell Ville, Córdoba | 2004 |
| Bab1 40 | 1 | leche | Morrison, Córdoba | 1996 |
| Bab1 41 | 1 | cepa | Río Cuarto, Córdoba | 2001 |
| Bab1 42 | 1 | feto bovino | Mercedes, Buenos Aires | 2005 |
| Bab1 43 | 1 | leche | V. Mackena, Córdoba | 2002 |
| Bab1 44 | 1 | leche | Buenos Aires | 1998 |
| Bab1 45 | 1 | s.i. | s.i. | s.i. |
| Bab1 46 | 1 | s.i. | s.i. | s.i. |
| Bab1 47 | 1 | s.i. | s.i. | s.i. |
| Bab1 48 | 1 | s.i. | s.i. | s.i. |
| Bab1 49 | 1 | s.i. | s.i. | s.i. |
| Bab1 50 | 1 | s.i. | s.i. | s.i. |
| Bab1 51 | 1 | s.i. | s.i. | s.i. |
| Bab2 1 | 2 | cepa | s.i. | 1994 |
| Bab2 2 | 2 | placenta | Luján, Buenos Aires | 1993 |
| Bab2 3 | 2 | leche | Los Toldos, Buenos Aires | 1983 |
| Bab2 4 | 2 | contenido estomacal | Chivilcoy, Buenos Aires | 1979 |
| Bab2 5 | 2 | feto bovino | Los Toldos, Buenos Aires | 1983 |
| Bab2 6 | 2 | leche | Luján, Buenos Aires | 1992 |
| Bab2 7 | 2 | leche | Los Toldos, Buenos Aires | 1983 |
La tipificación de cada cepa se realizó siguiendo las técnicas habituales1, según se detalla a continuación. Crecimiento en presencia de colorantes: las bacterias se sembraron en agar triptosa en presencia de diluciones 1: 50 000 de fucsina y 1: 25 000 y 1: 50 000 de tionina. Producción de H2S: las bacterias se sembraron en tubos con agar triptosa en pico de flauta, entre el tubo y el tapón se colocó una tira de papel seca, previamente embebida en una solución al 10 % de acetato de plomo. Las bacterias se dejaron crecer durante 4 días, con un cambio diario de la tira del papel indicador, en presencia de H2S dicho papel se oscurece. Prueba de ureasa: al medio de Christensen suplementado con urea se le agregó una ansada de cada cepa y se observó el cambio de color del medio, de amarillo a rosa. Cada cepa se sembró en agar triptosa (Difco, Sparks, EE.UU.) en pico de flauta y se dejó crecer en estufa a 37°C durante 48 h. Los cultivos de las cepas de la biovariedad 2 se suplementaron con 10 % de suero bovino negativo proveniente de animales de establecimientos libres de brucelosis y se dejaron crecer en atmósfera con 5 % de CO2. Luego las estrías se resuspendieron en 5 ml de NaCl 5 %. Las suspensiones se inactivaron a 82°C durante 60min. Se sembró una alícuota en agar triptosa según los requerimientos de cada cepa para verificar su inactivación y se dejó crecer en estufa a 37°C durante 72 h. Esta suspensión se utilizó como templado en las reacciones de PCR.
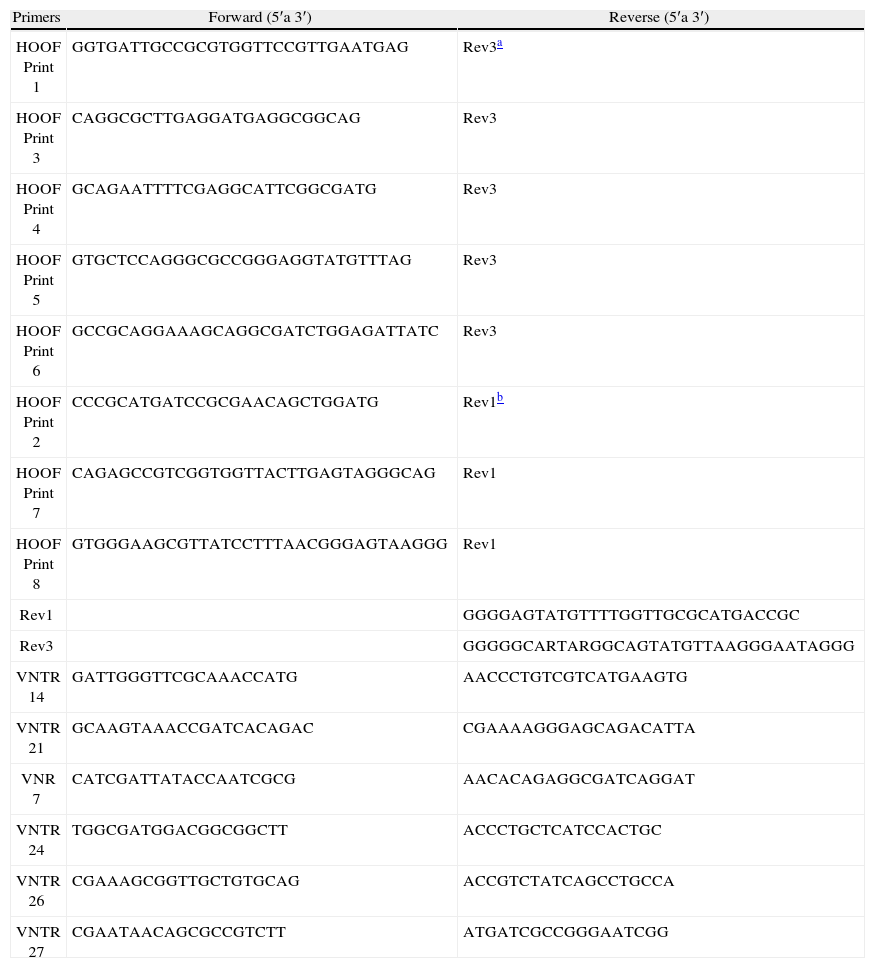
Amplificación por PCRLas reacciones de PCR se realizaron empleando la enzima Taq DNA Polymerase (Invitrogen, CA) en un termociclador Hybaid Px2 (Thermo Scientific, EE.UU.). Los oligonucleótidos utilizados (tabla 2) se adquirieron en Operon (EE.UU.). La amplifi- cación de los HOOF-Prints se realizó en las siguientes condiciones: 20mM Tris-HCl (pH 8,4), 50mM KCl, 0,25mM de cada dNTP, 2,5mM MgCl2, 0,2μM de cada oligonucleótido, 0,6 unidades de Taq y 3μl de templado. El programa de ciclado comprendió la siguiente secuencia: 94°C durante 2min, 32 ciclos de 94°C, 5 s; 55°C, 20 s y 72°C, 1,5min; con una extensión final a 72°C durante 5min. La amplificación de los VNTR se realizó en las siguientes condiciones: buffer de la enzima 1X, 0,25mM de cada dNTP, 2mM MgCl2, 0,2μM de cada oligonucleótido, 0,6 unidades de Taq y 1μl de templado. Las condiciones de ciclado fueron las siguientes: 95°C, 5min; 30 ciclos de 95°C, 30 s; 55°C, 30 s y 72°C, 1min; con una extensión final a 72°C durante 10min.
Oligonucleótidos utilizados en las reacciones de PCR.
| Primers | Forward (5′a 3′) | Reverse (5′a 3′) |
| HOOF Print 1 | GGTGATTGCCGCGTGGTTCCGTTGAATGAG | Rev3a |
| HOOF Print 3 | CAGGCGCTTGAGGATGAGGCGGCAG | Rev3 |
| HOOF Print 4 | GCAGAATTTTCGAGGCATTCGGCGATG | Rev3 |
| HOOF Print 5 | GTGCTCCAGGGCGCCGGGAGGTATGTTTAG | Rev3 |
| HOOF Print 6 | GCCGCAGGAAAGCAGGCGATCTGGAGATTATC | Rev3 |
| HOOF Print 2 | CCCGCATGATCCGCGAACAGCTGGATG | Rev1b |
| HOOF Print 7 | CAGAGCCGTCGGTGGTTACTTGAGTAGGGCAG | Rev1 |
| HOOF Print 8 | GTGGGAAGCGTTATCCTTTAACGGGAGTAAGGG | Rev1 |
| Rev1 | GGGGAGTATGTTTTGGTTGCGCATGACCGC | |
| Rev3 | GGGGGCARTARGGCAGTATGTTAAGGGAATAGGG | |
| VNTR 14 | GATTGGGTTCGCAAACCATG | AACCCTGTCGTCATGAAGTG |
| VNTR 21 | GCAAGTAAACCGATCACAGAC | CGAAAAGGGAGCAGACATTA |
| VNR 7 | CATCGATTATACCAATCGCG | AACACAGAGGCGATCAGGAT |
| VNTR 24 | TGGCGATGGACGGCGGCTT | ACCCTGCTCATCCACTGC |
| VNTR 26 | CGAAAGCGGTTGCTGTGCAG | ACCGTCTATCAGCCTGCCA |
| VNTR 27 | CGAATAACAGCGCCGTCTT | ATGATCGCCGGGAATCGG |
De los 13 loci VNTR descritos por Whatmore et al.24, se amplificaron VNTR 7, 14, 21, 24, 26 y 27. Los productos de amplificación se diluyeron 1:10 con agua y 5μl de la dilución se sembraron en geles de poliacrilamida al 8 % (Invitrogen, CA). A los geles se les agregó 1,5 % de glicerol para disminuir la difusión y evitar que se sequen por evaporación durante su conservación. Los geles de poliacrilamida se tiñeron con plata según el siguiente procedimiento: fijación con una solución con 10 % de etanol (Merck, Argentina) y 0,5 % de ácido acético (Anedra, Argentina) durante 10min, tinción con una solución al 0,2 % de AgNO3 (Merck, Argentina) durante 10min, un lavado con agua durante 5min, revelado con una solución con 3 % de NaOH (Mallinckrodt, EE.UU.) y 0,6 % de formol, hasta aparición de las bandas, y fijación durante 5min con la solución del primer paso. Luego se colocaron entre dos hojas de papel celofán y se dejaron secar a temperatura ambiente. Para determinar el tamaño de las bandas se utilizaron los marcadores de 100 bp DNA Ladder (Invitrogen, CA) y O′RangeRuler 20 bp DNA Ladder (Fermentas, Vilna, Lituania).
Análisis de la relación genéticaCon la herramienta V-DICE disponible en el sitio web de HPA (http://www.hpa-bioinformatics.org.uk/cgi-bin/DICI/DICI. pl) se determinó el poder de discriminación, utilizando el índice de discriminación de Hunter-Gaston (IDHG). El análisis de la relación genética entre los aislamientos se realizó mediante el algoritmo goeBURST, utilizando el software Phyloviz (http://www.phyloviz.net/wiki/)5.
ResultadosSe tipificaron las cepas por los métodos convencionales. Las cepas de la biovariedad 1 no requieren suero, no crecen en medio con tionina pero sí con fucsina, presentan actividad ureasa y producción de H2S. Las de la biovariedad 2 requieren suero, no crecen en medio con colorantes y también presentan actividad ureasa y producción de H2S. De acuerdo a las características bioquímicas evaluadas, 54 de las 61 cepas de B. abortus analizadas pertenecen a la biovariedad 1 y 7 a la biovariedad 2. Entre las cepas de la biovariedad 1 se encuentran la cepa de referencia 2308 y las cepas vacunales S19 y RB51.
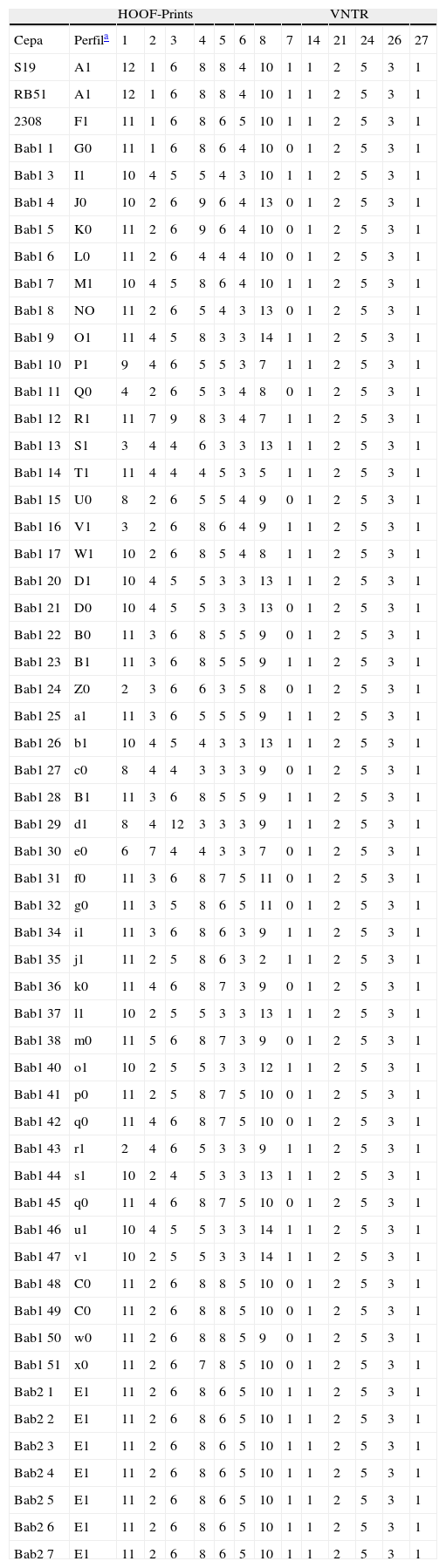
En la tabla 3 se muestran los perfiles genotípicos de los aislamientos de B. abortus para 7 marcadores HOOF-Prints y 6 marcadores VNTR. El poder resolutivo del esquema MLVA utilizado es muy alto, de los 56 perfiles genotípicos determinados, 47 son únicos. No se obtuvo producto de amplificación del HOOF-Print 7; Bricker et al.3 mencionan que esta amplifi cación se puede optimizar mediante PCR multiplex con los HOOF-Prints 2 y 8, pero en nuestro caso tampoco se logró de esa manera. Los perfiles HOOF-Prints obtenidos para las cepas S19 y RB51 difieren de aquellos publicados previamente24. Como era de esperar, ya que todos los aislamientos son de la misma especie, los loci VNTR analizados son monomórfi cos, excepto uno, el VNTR 7, que presenta dos alelos.
Perfi les genotípicos de los aislamientos.
| HOOF-Prints | VNTR | |||||||||||||
| Cepa | Perfila | 1 | 2 | 3 | 4 | 5 | 6 | 8 | 7 | 14 | 21 | 24 | 26 | 27 |
| S19 | A1 | 12 | 1 | 6 | 8 | 8 | 4 | 10 | 1 | 1 | 2 | 5 | 3 | 1 |
| RB51 | A1 | 12 | 1 | 6 | 8 | 8 | 4 | 10 | 1 | 1 | 2 | 5 | 3 | 1 |
| 2308 | F1 | 11 | 1 | 6 | 8 | 6 | 5 | 10 | 1 | 1 | 2 | 5 | 3 | 1 |
| Bab1 1 | G0 | 11 | 1 | 6 | 8 | 6 | 4 | 10 | 0 | 1 | 2 | 5 | 3 | 1 |
| Bab1 3 | I1 | 10 | 4 | 5 | 5 | 4 | 3 | 10 | 1 | 1 | 2 | 5 | 3 | 1 |
| Bab1 4 | J0 | 10 | 2 | 6 | 9 | 6 | 4 | 13 | 0 | 1 | 2 | 5 | 3 | 1 |
| Bab1 5 | K0 | 11 | 2 | 6 | 9 | 6 | 4 | 10 | 0 | 1 | 2 | 5 | 3 | 1 |
| Bab1 6 | L0 | 11 | 2 | 6 | 4 | 4 | 4 | 10 | 0 | 1 | 2 | 5 | 3 | 1 |
| Bab1 7 | M1 | 10 | 4 | 5 | 8 | 6 | 4 | 10 | 1 | 1 | 2 | 5 | 3 | 1 |
| Bab1 8 | NO | 11 | 2 | 6 | 5 | 4 | 3 | 13 | 0 | 1 | 2 | 5 | 3 | 1 |
| Bab1 9 | O1 | 11 | 4 | 5 | 8 | 3 | 3 | 14 | 1 | 1 | 2 | 5 | 3 | 1 |
| Bab1 10 | P1 | 9 | 4 | 6 | 5 | 5 | 3 | 7 | 1 | 1 | 2 | 5 | 3 | 1 |
| Bab1 11 | Q0 | 4 | 2 | 6 | 5 | 3 | 4 | 8 | 0 | 1 | 2 | 5 | 3 | 1 |
| Bab1 12 | R1 | 11 | 7 | 9 | 8 | 3 | 4 | 7 | 1 | 1 | 2 | 5 | 3 | 1 |
| Bab1 13 | S1 | 3 | 4 | 4 | 6 | 3 | 3 | 13 | 1 | 1 | 2 | 5 | 3 | 1 |
| Bab1 14 | T1 | 11 | 4 | 4 | 4 | 5 | 3 | 5 | 1 | 1 | 2 | 5 | 3 | 1 |
| Bab1 15 | U0 | 8 | 2 | 6 | 5 | 5 | 4 | 9 | 0 | 1 | 2 | 5 | 3 | 1 |
| Bab1 16 | V1 | 3 | 2 | 6 | 8 | 6 | 4 | 9 | 1 | 1 | 2 | 5 | 3 | 1 |
| Bab1 17 | W1 | 10 | 2 | 6 | 8 | 5 | 4 | 8 | 1 | 1 | 2 | 5 | 3 | 1 |
| Bab1 20 | D1 | 10 | 4 | 5 | 5 | 3 | 3 | 13 | 1 | 1 | 2 | 5 | 3 | 1 |
| Bab1 21 | D0 | 10 | 4 | 5 | 5 | 3 | 3 | 13 | 0 | 1 | 2 | 5 | 3 | 1 |
| Bab1 22 | B0 | 11 | 3 | 6 | 8 | 5 | 5 | 9 | 0 | 1 | 2 | 5 | 3 | 1 |
| Bab1 23 | B1 | 11 | 3 | 6 | 8 | 5 | 5 | 9 | 1 | 1 | 2 | 5 | 3 | 1 |
| Bab1 24 | Z0 | 2 | 3 | 6 | 6 | 3 | 5 | 8 | 0 | 1 | 2 | 5 | 3 | 1 |
| Bab1 25 | a1 | 11 | 3 | 6 | 5 | 5 | 5 | 9 | 1 | 1 | 2 | 5 | 3 | 1 |
| Bab1 26 | b1 | 10 | 4 | 5 | 4 | 3 | 3 | 13 | 1 | 1 | 2 | 5 | 3 | 1 |
| Bab1 27 | c0 | 8 | 4 | 4 | 3 | 3 | 3 | 9 | 0 | 1 | 2 | 5 | 3 | 1 |
| Bab1 28 | B1 | 11 | 3 | 6 | 8 | 5 | 5 | 9 | 1 | 1 | 2 | 5 | 3 | 1 |
| Bab1 29 | d1 | 8 | 4 | 12 | 3 | 3 | 3 | 9 | 1 | 1 | 2 | 5 | 3 | 1 |
| Bab1 30 | e0 | 6 | 7 | 4 | 4 | 3 | 3 | 7 | 0 | 1 | 2 | 5 | 3 | 1 |
| Bab1 31 | f0 | 11 | 3 | 6 | 8 | 7 | 5 | 11 | 0 | 1 | 2 | 5 | 3 | 1 |
| Bab1 32 | g0 | 11 | 3 | 5 | 8 | 6 | 5 | 11 | 0 | 1 | 2 | 5 | 3 | 1 |
| Bab1 34 | i1 | 11 | 3 | 6 | 8 | 6 | 3 | 9 | 1 | 1 | 2 | 5 | 3 | 1 |
| Bab1 35 | j1 | 11 | 2 | 5 | 8 | 6 | 3 | 2 | 1 | 1 | 2 | 5 | 3 | 1 |
| Bab1 36 | k0 | 11 | 4 | 6 | 8 | 7 | 3 | 9 | 0 | 1 | 2 | 5 | 3 | 1 |
| Bab1 37 | l1 | 10 | 2 | 5 | 5 | 3 | 3 | 13 | 1 | 1 | 2 | 5 | 3 | 1 |
| Bab1 38 | m0 | 11 | 5 | 6 | 8 | 7 | 3 | 9 | 0 | 1 | 2 | 5 | 3 | 1 |
| Bab1 40 | o1 | 10 | 2 | 5 | 5 | 3 | 3 | 12 | 1 | 1 | 2 | 5 | 3 | 1 |
| Bab1 41 | p0 | 11 | 2 | 5 | 8 | 7 | 5 | 10 | 0 | 1 | 2 | 5 | 3 | 1 |
| Bab1 42 | q0 | 11 | 4 | 6 | 8 | 7 | 5 | 10 | 0 | 1 | 2 | 5 | 3 | 1 |
| Bab1 43 | r1 | 2 | 4 | 6 | 5 | 3 | 3 | 9 | 1 | 1 | 2 | 5 | 3 | 1 |
| Bab1 44 | s1 | 10 | 2 | 4 | 5 | 3 | 3 | 13 | 1 | 1 | 2 | 5 | 3 | 1 |
| Bab1 45 | q0 | 11 | 4 | 6 | 8 | 7 | 5 | 10 | 0 | 1 | 2 | 5 | 3 | 1 |
| Bab1 46 | u1 | 10 | 4 | 5 | 5 | 3 | 3 | 14 | 1 | 1 | 2 | 5 | 3 | 1 |
| Bab1 47 | v1 | 10 | 2 | 5 | 5 | 3 | 3 | 14 | 1 | 1 | 2 | 5 | 3 | 1 |
| Bab1 48 | C0 | 11 | 2 | 6 | 8 | 8 | 5 | 10 | 0 | 1 | 2 | 5 | 3 | 1 |
| Bab1 49 | C0 | 11 | 2 | 6 | 8 | 8 | 5 | 10 | 0 | 1 | 2 | 5 | 3 | 1 |
| Bab1 50 | w0 | 11 | 2 | 6 | 8 | 8 | 5 | 9 | 0 | 1 | 2 | 5 | 3 | 1 |
| Bab1 51 | x0 | 11 | 2 | 6 | 7 | 8 | 5 | 10 | 0 | 1 | 2 | 5 | 3 | 1 |
| Bab2 1 | E1 | 11 | 2 | 6 | 8 | 6 | 5 | 10 | 1 | 1 | 2 | 5 | 3 | 1 |
| Bab2 2 | E1 | 11 | 2 | 6 | 8 | 6 | 5 | 10 | 1 | 1 | 2 | 5 | 3 | 1 |
| Bab2 3 | E1 | 11 | 2 | 6 | 8 | 6 | 5 | 10 | 1 | 1 | 2 | 5 | 3 | 1 |
| Bab2 4 | E1 | 11 | 2 | 6 | 8 | 6 | 5 | 10 | 1 | 1 | 2 | 5 | 3 | 1 |
| Bab2 5 | E1 | 11 | 2 | 6 | 8 | 6 | 5 | 10 | 1 | 1 | 2 | 5 | 3 | 1 |
| Bab2 6 | E1 | 11 | 2 | 6 | 8 | 6 | 5 | 10 | 1 | 1 | 2 | 5 | 3 | 1 |
| Bab2 7 | E1 | 11 | 2 | 6 | 8 | 6 | 5 | 10 | 1 | 1 | 2 | 5 | 3 | 1 |
El código de cada perfi l correspondiente a la combinación de HOOF-Print y VNTR está formado por una letra y un número. Cada perfi l diferente obtenido para los HOOF-Prints se identifi có con una letra del alfabeto (la misma letra en mayúscula o minúscula indica perfi les diferentes). Los dos perfi les obtenidos para los VNTR se designaron 0 y 1.
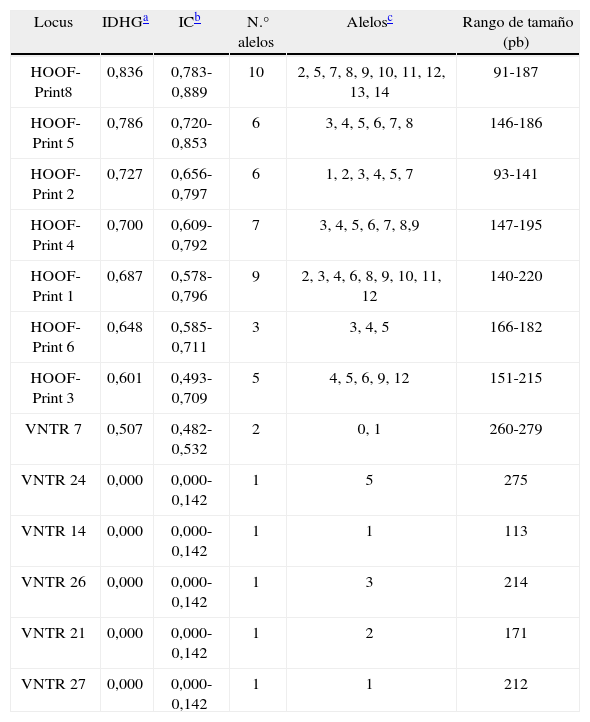
El número de alelos observado en cada locus en la población analizada está entre 1, en el caso de los VNTR, y 10, en el caso del HOOF-Print 8. Las frecuencias de los alelos HOOF- Prints se puede observar en la figura 1 Se calculó el índice de discriminación (ID) de cada locus según el índice de Hunter-Gaston (tabla 4), como sugiere el ESGEM.

Poder discriminatorio de los 13 loci analizados en este trabajo.
| Locus | IDHGa | ICb | N.° alelos | Alelosc | Rango de tamaño (pb) |
| HOOF-Print8 | 0,836 | 0,783-0,889 | 10 | 2, 5, 7, 8, 9, 10, 11, 12, 13, 14 | 91-187 |
| HOOF-Print 5 | 0,786 | 0,720-0,853 | 6 | 3, 4, 5, 6, 7, 8 | 146-186 |
| HOOF-Print 2 | 0,727 | 0,656-0,797 | 6 | 1, 2, 3, 4, 5, 7 | 93-141 |
| HOOF-Print 4 | 0,700 | 0,609-0,792 | 7 | 3, 4, 5, 6, 7, 8,9 | 147-195 |
| HOOF-Print 1 | 0,687 | 0,578-0,796 | 9 | 2, 3, 4, 6, 8, 9, 10, 11, 12 | 140-220 |
| HOOF-Print 6 | 0,648 | 0,585-0,711 | 3 | 3, 4, 5 | 166-182 |
| HOOF-Print 3 | 0,601 | 0,493-0,709 | 5 | 4, 5, 6, 9, 12 | 151-215 |
| VNTR 7 | 0,507 | 0,482-0,532 | 2 | 0, 1 | 260-279 |
| VNTR 24 | 0,000 | 0,000-0,142 | 1 | 5 | 275 |
| VNTR 14 | 0,000 | 0,000-0,142 | 1 | 1 | 113 |
| VNTR 26 | 0,000 | 0,000-0,142 | 1 | 3 | 214 |
| VNTR 21 | 0,000 | 0,000-0,142 | 1 | 2 | 171 |
| VNTR 27 | 0,000 | 0,000-0,142 | 1 | 1 | 212 |
De los aislamientos presentados en la tabla 1, tres poseen múltiples alelos para un locus dado (Bab1 2, 18 y 19) y de dos de ellos (Bab1 33 y 39) no se logró amplificar el alelo del HOOF-Print 8, por lo tanto, los cinco se excluyeron del estudio. La relación entre variantes genéticas descritas a partir de los 56 aislamientos restantes se determinó mediante el algoritmo goeBURST. Este algoritmo construye complejos clonales compuestos por aquellos genotipos relacionados entre sí y que solo varían en 1 a 3 marcadores del set de 13 estudiados. Los genotipos descritos en el presente estudio se relacionaron en un gran complejo clonal, dentro del cual se pueden observar tres grandes agrupamientos conectados por el genotipo E1, correspondiente a los aislamientos de la biovariedad 2 (fig. 2A). Así, E1 correspondería al genotipo fundador del gran complejo clonal definido según variantes triples de alelos.
. Cada círculo representa un único perfi l, su tamaño refl eja la frecuencia de ese genotipo. Los círculos rojos corresponden a aislamientos de la biovariedad 1 y los azules a la biovariedad 2. El número sobre las ramas indica en cuántos loci difi eren los dos aislamientos conectados. B. Relación geográfi ca entre los perfi les genotípicos de los aislamientos. Los colores de los círculos representan a las diferentes provincias y los números indican la localidad de dónde proviene la cepa. C. Mapa de la Argentina donde están indicados, en la región ampliada, los lugares de procedencia de los aislamientos analizados. La mayoría de los aislamientos provienen del sur de Córdoba y del noreste de Buenos Aires.")
Relación entre los aislamientos y la cepa de referencia mediante goeBURST a partir de los 13 marcadores estudiados. F1 corresponde al genotipo de la cepa de referencia B. abortus 2308. A. Se observan un complejo clonal con el genotipo fundador E1. El genotipo F1 corresponde a la cepa 2308 de la cual derivan las cepas vacunales S19 y RB51 (genotipo A1). Cada círculo representa un único perfi l, su tamaño refl eja la frecuencia de ese genotipo. Los círculos rojos corresponden a aislamientos de la biovariedad 1 y los azules a la biovariedad 2. El número sobre las ramas indica en cuántos loci difi eren los dos aislamientos conectados. B. Relación geográfi ca entre los perfi les genotípicos de los aislamientos. Los colores de los círculos representan a las diferentes provincias y los números indican la localidad de dónde proviene la cepa. C. Mapa de la Argentina donde están indicados, en la región ampliada, los lugares de procedencia de los aislamientos analizados. La mayoría de los aislamientos provienen del sur de Córdoba y del noreste de Buenos Aires.
Por otro lado, el genotipo A1 (cepas vacunales) se encontró altamente relacionado con el F1, el cual corresponde a la cepa a partir de la cual se obtuvieron las cepas vacunales (B. abortus 2308). El algoritmo goeBURST permite realizar un análisis de los complejos clonales según datos asociados a la colección de muestras genotipificadas. De esta manera, los complejos clonales se evaluaron en el contexto espacial y temporal de la colección en estudio. Teniendo en cuenta el lugar de procedencia de las cepas, el análisis no mostró correlación entre la procedencia geográfica y los genotipos (figs. 2B y C), mientras que resultó interesante observar que el genotipo fundador E1 no solo fue el más frecuente, sino que además pudo ser aislado en diversos puntos en el tiempo (resultado no mostrado).
DiscusiónEl esquema de MLVA utilizado en este trabajo se eligió sobre la base de los esquemas publicados al momento de diseñar este estudio y la habilidad de ellos para tipificar los aislamientos, así como de la disponibilidad de recursos y tecnología y de la experiencia requerida para realizar la técnica.
Como los marcadores HOOF-Prints son muy discriminatorios, la gran mayoría de los perfiles obtenidos resultaron ser únicos. La discrepancia observada entre los perfiles HOOFPrints publicados y la obtenida en ese trabajo para las cepas vacunales S19 y RB51 se debe a la alta tasa de mutación de los loci analizados. De hecho, el polimorfismo observado en las regiones no codificantes del genoma donde se encuentran unidades repetidas en tándem se debe a la alta tasa de mutación de estos loci. Pero, por otro lado, esto también dificulta la asignación de un aislamiento a una de las especies reconocidas. Los HOOF-Prints son muy eficientes para distinguir cepas en un brote local, pero son incapaces de predecir correctamente la biovariedad o la especie a la que corresponde un aislamiento dado11. Es por eso que el análisis de los resultados debe realizarse con cautela y utilizar la genotipificación por los HOOF-Prints como complemento de los estudios epidemiológicos tradicionales, no como único método de tipificación.
Cuando se compara el poder discriminatorio de los HOOFPrints obtenidos en este trabajo con los disponibles en la literatura, no hay diferencias importantes, excepto en el HOOF-Print 8. En el presente trabajo se obtuvo un IDHG de 0,83, mientras que en el estudio de Bricker et al.4 dicho valor fue 0. Esta discrepancia puede deberse a los criterios utilizados para la selección de los aislamientos en estudio. Dado que la diversidad global de los VNTR utilizados por Whatmore et al. es similar a la de los HOOF-Prints para una misma población, poseen el mismo poder de discriminación24.
Las dificultades técnicas ocasionadas al tener 7 loci adicionales de difícil detección para nuestras condiciones de trabajo no traerían aparejado un aumento significativo en el poder de discriminación del método, por este motivo se decidió que el grupo de marcadores hipervariables estaría compuesto únicamente por los loci HOOF-Prints, y el grupo de marcadores más estables lo formarían los loci VNTR seleccionados. Los loci VNTR empleados en este trabajo presentaron escasa o nula variabilidad ya que fueron seleccionados con el objeto de diferenciar las distintas especies. Se los incluyó en este estudio para comparar el perfil genotípico de este conjunto de aislamientos locales con aquellos publicados por otros autores para cepas de B. abortus de otras regiones del mundo, y sorprendentemente, se encontraron diferencias. En el trabajo de Whatmore et al.24, el alelo de las cepas de B. abortus para el locus VNTR 7 es el 2 y para el locus VNTR 26 es el 4, mientras que en el presente trabajo los alelos obtenidos fueron 0 y 1 para el locus VNTR 7, y 1 para el locus VNTR 26. Si bien estos marcadores son muy estables, no están exentos de sufrir mutaciones; de hecho, los autores mencionados anteriormente encontraron una deleción de 3 pb en una de las copias de la repetición en un alelo del VNTR 724.
Existen en la literatura ejemplos del uso combinado de un grupo de marcadores HOOF-Prints y VNTR. El esquema de 10 loci VNTR utilizado por Beja-Pereira et al.2 y Higgins et al.8 está compuesto por una combinación de 4 marcadores HOOF-Prints y 6 VNTR, que surgió de un estudio llevado a cabo por B. Harris en 20088. A partir de la genotipificación de 82 aislamientos de Brucella utilizando el esquema completo de 21 marcadores, este autor eligió aquellos 10 que permiten diferenciar los aislamientos en clusters relevantes desde el punto de vista epidemiológico.
Según los resultados obtenidos en este trabajo, el genotipo E1 se encuentra únicamente en la provincia de Buenos Aires. Pero considerando que la mayoría de los aislamientos provienen de dicha provincia, esta distribución podría deberse a un sesgo de la muestra.
Por otro lado, es posible que la rápida evolución de los loci Hoof-Prints enmascare las diferencias regionales en la composición de los genotipos, es decir, se podrían obtener dos genotipos iguales en aislamientos no relacionados. La destacable permanencia temporal del genotipo E1 indicaría que estos aislamientos tendrían una alta eficacia biológica (fitness). Aún cuando el genotipo E1 fuera el más frecuente, solo por un hecho matemático, esto aumentaría la probabilidad de que sufra mutaciones y que, de esta manera, se generen nuevas cepas y/o especies.
Este trabajo aporta evidencias a favor de la teoría de Margaret Meyer17, quien, basándose en evidencias bioquímicas, en la década de los 80 postuló que sería la biovariedad 2 de B. abortus el organismo que dio origen a todo el género.
Como herramienta epidemiológica, el MLVA ofrece una destacable resolución comparado con los métodos de tipifi- cación convencionales, que generalmente no tienen poder discriminativo por debajo del nivel de biovariedad. Los marcadores que se incluyen en un estudio de MLVA de forma separada no son informativos, son muy variables o muestran un alto nivel de homoplasia, pero la combinación de un grupo bien seleccionado de estos marcadores podría ser muy discriminativa. Actualmente se está empleando el esquema de MLVA propuesto por Le Flèche et al.11; los agrupamientos de las cepas de B. abortus en biovariedades 1, 2 y 4 generados por este esquema concuerdan con los resultados de la tipificación bioquímica y el patrón de PCR-RFLP para el gen omp2a. El MLVA es una técnica relativamente sencilla, ya que solo requiere determinar el tamaño del producto de amplificación de la PCR (que se puede hacer de manera automática, mediante electroforesis capilar en un secuenciador de ADN, o con los métodos de electroforesis tradicionales en geles de agarosa o poliacrilamida), y además permite el fácil intercambio de información entre grupos, por ejemplo, mediante la construcción de bases de datos on line con los perfiles genotípicos de todos los aislamientos, como MLVA Bank16.
En suma, el empleo de este esquema de MLVA en aislamientos de Brucella abortus de Argentina permitió establecer la biovariedad a la que estos corresponden. Asimismo, a través del análisis goeBURST se pudo relacionar a todos los genotipos entre sí y proponer al genotipo de la biovariedad 2 como fundador.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Este trabajo fue financiado por los proyectos de INTA «Brucelosis » (AESA 2581) y «Conservación y valoración de recursos genéticos microbiológicos» (AERG-231251). Los autores agradecen a la Dra. Paula Ruybal por su invaluable colaboración en el análisis goeBURST y por la lectura y corrección del manuscrito. Durante el desarrollo de este trabajo, DH era becaria de CONICET, Argentina.
