




El síndrome poliglandular autoinmune tipo 1 (SPA-1), también conocido como síndrome APECED (autoimmune-polyendocrinopathty-candidiasis-ectodermal-dystrophy, OMIM 240300) o síndrome de Whitaker es una entidad infrecuente, que se transmite mediante herencia autosómica recesiva. Se ha identificado el gen AIRE, localizado en el cromosoma 21, como responsable de la hipofunción glandular en estos pacientes.
Además de la afectación endocrina, se han descrito otras alteraciones autoinmunitarias no endocrinológicas (hepatitis autoinmune, gastritis atrófica, malabsorción intestinal) y anomalías ectodérmicas (hipoplasia del esmalte dentario, queratitis, distrofia ungueal). Para su diagnóstico, es preciso que aparezcan al menos dos de los siguientes criterios: candidiasis mucocutánea crónica (CMCC), hipoparatiroidismo e insuficiencia suprarrenal, o solo uno de ellos en presencia de un hermano afectado.
Muchos de los pacientes con SPA-1 desarrollarán patología cutánea, por lo que el dermatólogo debe conocer aquellas manifestaciones que pueden conducir a un diagnóstico precoz del síndrome.
IntroducciónEn 1849 Addison describió la asociación de anemia perniciosa e insuficiencia suprarrenal y a partir de entonces se comienzan a describir otras enfermedades de base autoinmune1. En 1929 se estableció la asociación entre hipoparatiroidismo y candidiasis mucocutánea y en 1946 se comunicó la relación de estas dos enfermedades con la insuficiencia suprarrenal. El término de síndrome poliglandular autoinmunitario se empezó a utilizar en 1980 para describir la asociación de insuficiencia suprarrenal primaria, tiroidopatía autoinmunitaria y candidiasis mucocutánea crónica2. Se definieron entonces dos tipos de síndromes poliglandulares autoinmunes: el tipo 1 asocia la insuficiencia suprarrenal con el hipoparatiroidsimo y el tipo 2 la insuficiencia suprarrenal con la enfermedad tiroidea y con la diabetes mellitus tipo 1. Estos síndromes pueden desarrollar otras alteraciones endocrinas y no endocrinas, pero de naturaleza autoinmune. Recientemente se ha descrito un síndrome poliglandular autoinmune tipo 3 en adultos, en el que, a diferencia de los tipos 1 y 2, no se produce afectación de las glándulas suprarrenales. Esta sería la única diferencia entre los tipo 2 y 33–6.
EpidemiologíaEl SPA-1 es una entidad infrecuente. Sin embargo, la frecuencia es mayor en poblaciones pequeñas o con un alto grado de consanguineidad. Así, en Finlandia se ha descrito una prevalencia de 1:25.0002, en judíos iraníes de 1:9.0004 y en sardos de 1:14.5001. Por el contrario, en otras poblaciones, la incidencia es mucho más baja, como ocurre en Noruega (1:80.000)7,8. Parece que existe un ligero predominio femenino de la enfermedad, aunque los datos varían según las series (ratio mujer/hombre entre 2,4/1 y 0,8/1)3,9. La enfermedad suele iniciarse ya en la infancia y el espectro de manifestaciones es muy variado en cada paciente.
Clásicamente se han utilizado criterios clínicos para su diagnóstico. La identificación del gen AIRE (autoimmune regulator) en 1997 como responsable de la enfermedad ha servido para establecer el diagnóstico definitivo y detectar nuevos casos precozmente, antes de que la enfermedad se manifieste de forma florida10,11.
Genética y autoinmunidadEl gen AIRE se localiza en el cromosoma 21q22.3 y se compone de 14 exones, que codifican para la proteína AIRE, de 545 aminoácidos y un peso molecular de 57,7kDa12. Este gen se expresa principalmente en el timo, en los ganglios linfáticos, en el hígado fetal y en sangre periférica (monocitos y células dendríticas), por lo que resulta de gran importancia para el correcto funcionamiento del sistema inmunitario7-13. En el SPA-1 se han descrito más de 60 mutaciones en este gen, número que se incrementa periódicamente en la literatura14-16. Las más frecuentes son la mutación c.769C>T (p.Arg257Stop) en la población finlandesa y la c.967_979del13 (p.Cys322fs) en las poblaciones noruega, norteamericana, inglesa, irlandesa y del noroeste francés7,17.
En este síndrome no se ha visto asociación con el sistema de los antígenos leucocitarios humanos. Dentro de una familia, suele afectarse una misma generación, pudiendo desarrollarse el síndrome en varios miembros, sin afectación de la generación posterior. Las manifestaciones son variables dentro de cada familia.
El gen AIRE juega un papel importante en el establecimiento de la tolerancia inmunológica central y periférica. El producto codificado se encuentra en el núcleo celular e interviene en la formación de múltiples proteínas encargadas de la regulación transcripcional. Las proteínas codificadas por el gen AIRE participan en la selección negativa de linfocitos T. Son responsables de que las poblaciones autorreactivas de células T sean eliminadas, lo que impide su migración a la periferia y el desarrollo posterior de manifestaciones autoinmunes18. En los ratones Aire-/-, esta selección negativa no se produce. Por tanto, los clones de células T autorreactivas migran hacia la periferia y son la causa de las enfermedades autoinmunes del tejido específico contra el que van dirigidas.
En las células del estroma del bazo y en los ganglios linfáticos, el gen AIRE controla la expresión de múltiples proteínas implicadas en la tolerancia inmunológica periférica. Esas proteínas son las encargadas de la eliminación de células T autorreactivas que han conseguido escapar previamente de la selección negativa del timo y juegan un papel muy relevante en la tolerancia inmune periférica19-21.
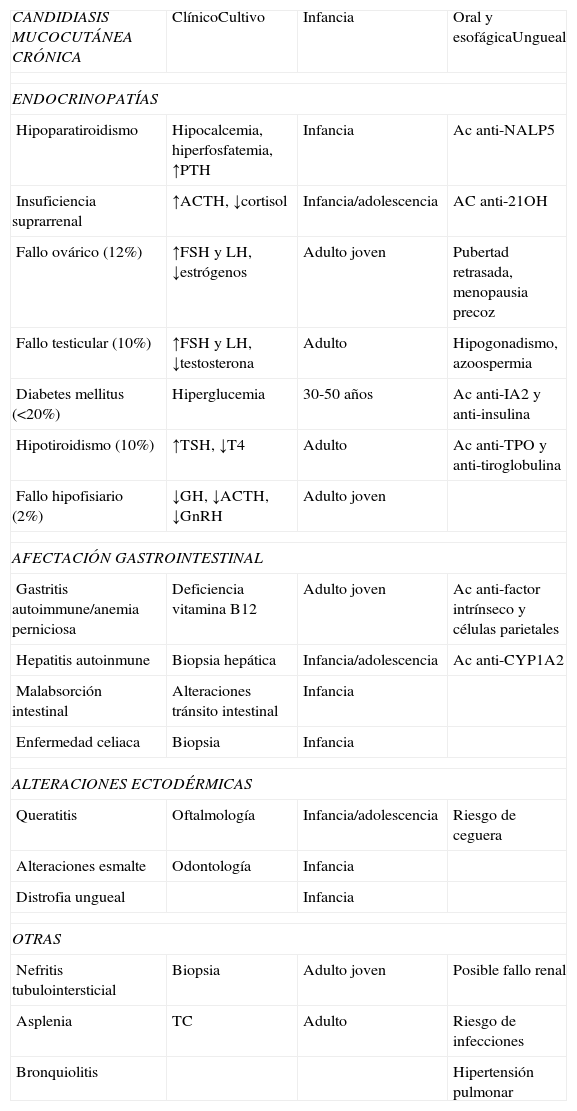
Criterios diagnósticos y presentación clínicaLa destrucción progresiva de los tejidos glandulares endocrinos desencadena manifestaciones variables, que incluyen hipoparatiroidismo, insuficiencia suprarrenal, hipotiroidismo, hipogonadismo o diabetes mellitus insulinodependiente, entre muchas otras (tabla 1).
Manifestaciones clínicas en los pacientes con SPA-1
| CANDIDIASIS MUCOCUTÁNEA CRÓNICA | ClínicoCultivo | Infancia | Oral y esofágicaUngueal |
| ENDOCRINOPATÍAS | |||
| Hipoparatiroidismo | Hipocalcemia, hiperfosfatemia, ↑PTH | Infancia | Ac anti-NALP5 |
| Insuficiencia suprarrenal | ↑ACTH, ↓cortisol | Infancia/adolescencia | AC anti-21OH |
| Fallo ovárico (12%) | ↑FSH y LH, ↓estrógenos | Adulto joven | Pubertad retrasada, menopausia precoz |
| Fallo testicular (10%) | ↑FSH y LH, ↓testosterona | Adulto | Hipogonadismo, azoospermia |
| Diabetes mellitus (<20%) | Hiperglucemia | 30-50 años | Ac anti-IA2 y anti-insulina |
| Hipotiroidismo (10%) | ↑TSH, ↓T4 | Adulto | Ac anti-TPO y anti-tiroglobulina |
| Fallo hipofisiario (2%) | ↓GH, ↓ACTH, ↓GnRH | Adulto joven | |
| AFECTACIÓN GASTROINTESTINAL | |||
| Gastritis autoimmune/anemia perniciosa | Deficiencia vitamina B12 | Adulto joven | Ac anti-factor intrínseco y células parietales |
| Hepatitis autoinmune | Biopsia hepática | Infancia/adolescencia | Ac anti-CYP1A2 |
| Malabsorción intestinal | Alteraciones tránsito intestinal | Infancia | |
| Enfermedad celiaca | Biopsia | Infancia | |
| ALTERACIONES ECTODÉRMICAS | |||
| Queratitis | Oftalmología | Infancia/adolescencia | Riesgo de ceguera |
| Alteraciones esmalte | Odontología | Infancia | |
| Distrofia ungueal | Infancia | ||
| OTRAS | |||
| Nefritis tubulointersticial | Biopsia | Adulto joven | Posible fallo renal |
| Asplenia | TC | Adulto | Riesgo de infecciones |
| Bronquiolitis | Hipertensión pulmonar | ||
La tríada clásica del SPA-1 incluye la CMCC, hipoparatiroidismo e insuficiencia suprarrenal. Estos tres componentes aparecen en un preciso orden cronológico, pero los tres, de forma conjunta, solo están presentes en el 35-50% de los pacientes22,23. Cuanto menor sea la edad de aparición del primero de estos componentes, con mayor facilidad se desarrollarán el resto, mientras que en aquellos pacientes en los que las manifestaciones aparezcan más tardíamente, menor afectación clínica tendrán.
La CMCC es la primera manifestación de este síndrome y suele aparecer antes de los 5 años de edad9,24.
El hipoparatiroidismo es la primera alteración endocrinológica en aparecer9. Suele manifestarse antes de los 10 años de edad, oscilando entre los 3 meses hasta los 44 años. La mediana de edad de aparición es a los 9 años y afecta al 73-100% de los pacientes2,3,9. Es más frecuente en las mujeres que en los hombres25. Se manifiesta por hipocalcemia junto con hiperfostatemia y niveles normales o disminuidos de la hormona paratiroidea (PTH) y con una función renal preservada. El paciente puede presentar síntomas de hipocalcemia, manifestados en forma de calambres musculares, parestesias o torpeza. En otras ocasiones, una infección o una situación estresante pueden desencadenar una hipocalcemia brusca, que se manifiesta en forma de crisis convulsivas. Recientemente se ha identificado a la proteína NALP5 (NACHT leucine-reich repeat protein 5) de las células paratiroides como la diana inmunológica de los autoanticuerpos responsables de la destrucción glandular26.
La insuficiencia suprarrenal primaria suele ser el último componente de la tríada en aparecer9. La edad de inicio oscila entre los 5 y los 15 años, con una prevalencia del 40% a los 10 años de edad y del 78% a los 30 años en una serie de pacientes finlandeses27. Se manifiesta por fatiga, avidez por la sal, hipotensión, pérdida de peso e hiperpigmentación de la piel y las mucosas. Se detectan niveles bajos de cortisol y aumentados de la hormona adrenocorticotrofa (ACTH). Como en otras adrenalitis autoinmunes, el principal antígeno contra el que van dirigidos los anticuerpos es la enzima 21-hidroxilasa, pero se han detectado también autoanticuerpos contra el citocromo p450 con escisión de la cadena lateral (citP450scc) (side chain cleavage) y contra la L-aminoácido aromático decarboxilasa28,29.
La destrucción autoinmune de otros órganos no endocrinos puede desencadenar hepatitis autoinmune, anemia perniciosa, gastritis atrófica, hipoesplenia/asplenia, síndrome de malabsorción, bronquiolitis y déficits celulares y humorales.
Manifestaciones cutáneascandidiasis mucocutánea crónicaEl género Candida está constituido por hongos levaduriformes e incluye a un centenar de especies, de las que tan solo 8 se encuentran asociadas a infecciones humanas, siendo C. albicans y tropicalis responsables del 80% de los casos. Habitualmente estas levaduras son comensales o saprófitos en el ser humano, pero tienen un potencial significativo para desarrollar infecciones oportunistas. Las candidiasis superficiales aparecen con más frecuencia en localizaciones con alto grado de humedad, oclusión o en áreas con pérdida de integridad de la barrera cutánea. Afectan especialmente a pacientes con algún tipo de alteración inmunológica y son más frecuentes en los extremos de la vida y en pacientes diabéticos30.
Los pacientes con SPA-1 desarrollan infecciones por Candida ya en los primeros años de vida. Se considera que es una expresión clínica de un déficit selectivo de las células T ante la levadura. La respuesta de las células B frente a Candida es normal, lo que previene el desarrollo de una candidiasis sistémica3,31. En la mayoría de los casos la afectación no supera el 5% de la superficie corporal. Suele ser la primera manifestación clínica del SPA-1. La infección suele ser crónica o recurrente y es habitual la resistencia a los tratamientos convencionales. En una serie finlandesa, estaba presente en la mitad de los pacientes de un año de edad, en el 70% de los de 5 años, en el 94% a los 20 años y en el 97% a los 30 años27. Por este motivo, los niños con CMCC deben ser evaluados periódicamente para reconocer precozmente los síntomas de una hipofunción endocrinológica asociada.
La mayoría de los casos se inician en la infancia como una estomatitis candidósica crónica rebelde al tratamiento. En los primeros meses de vida puede aparecer una estomatitis aguda pseudomembranosa (muguet). Se forman placas blanquecinas en las mucosas yugal, lingual y en las encías y en el paladar. Estas placas pueden confluir ocupando casi toda la cavidad oral. Si se retira la capa blanquecina, se observa una mucosa roja, brillante y erosiva.
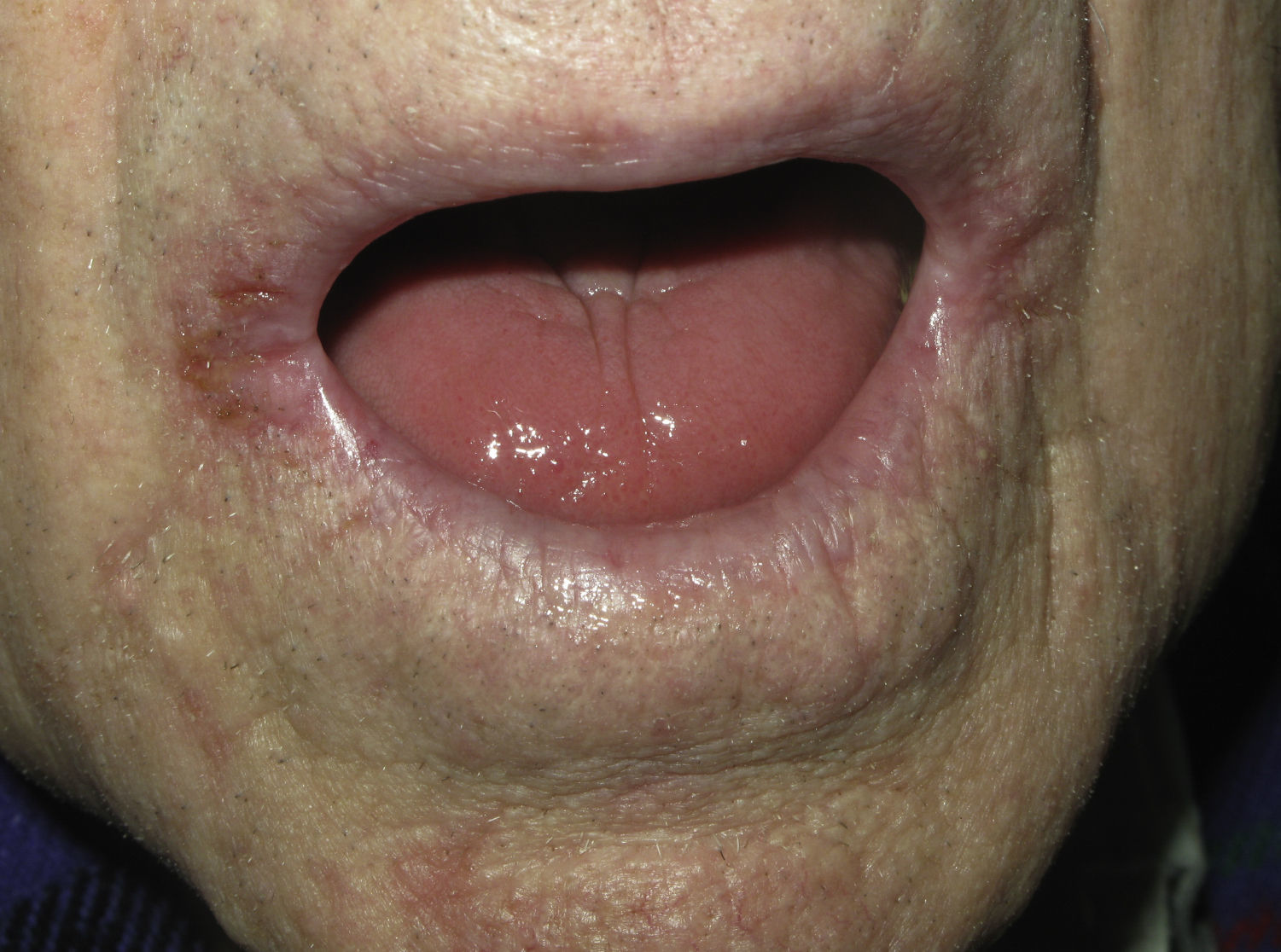
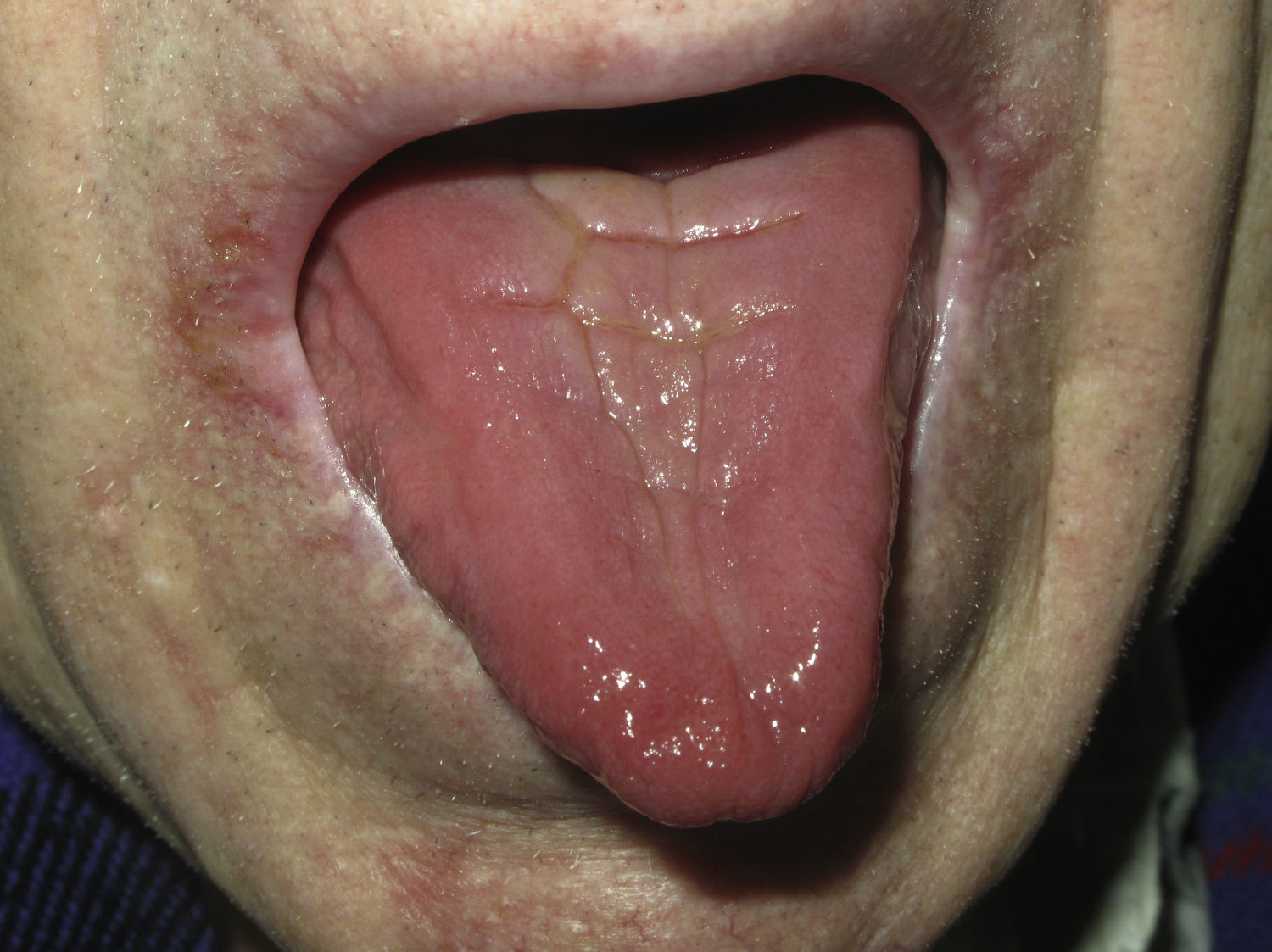
Posteriormente, los pacientes presentan lesiones labiales de tipo granulomatoso y una queilitis angular persistente. Esta forma se manifiesta como fisuras muy dolorosas en ambas comisuras bucales (fig. 1). Suele asociarse a otras formas de candidiasis oral, como la estomatitis candidósica atrófica. En estos casos el paciente refiere sensación de ardor y quemazón y se observa una lengua roja depapilada (fig. 2). También pueden desarrollar candidiasis esofágica y laríngea, que se manifiesta como dolor retroesternal e incluso como estenosis esofágica. En el resto del tubo digestivo, la candidiasis puede producir dolor abdominal, flatulencia y diarrea. En ocasiones existe afectación intestinal sin enfermedad oral3,9.


La inflamación repetida de las mucosas por la infección crónica predispone al desarrollo de tumores. Así, se ha descrito asociación con el carcinoma epidermoide de las mucosas oral y esofágica y con el adenocarcinoma gástrico27,32. La estomatitis candidósica atrófica se ha asociado a la aparición de carcinomas epidermoides con mayor frecuencia.
En los pacientes con SPA-1 también es frecuente el desarrollo de candidiasis genital y la afectación de pliegues y de uñas.
- •
Balanitis candidósica
En las formas leves aparece eritema y prurito poscoital de evolución transitoria. En las formas más graves se observa un glande eritematoso con pápulas y pústulas, que pueden evolucionar a una balanitis erosiva. Es posible la progresión a la piel del prepucio y pliegues.
- •
Vulvovaginitis candidósica
Se caracteriza por eritema, vesículas y pústulas en el introito vulvar, acompañado por un flujo vaginal blanquecino y espeso y sensación de escozor y prurito. Las recurrencias son frecuentes al ser Candida un colonizador habitual del tracto orointestinal.
- •
Intertrigo candidósico
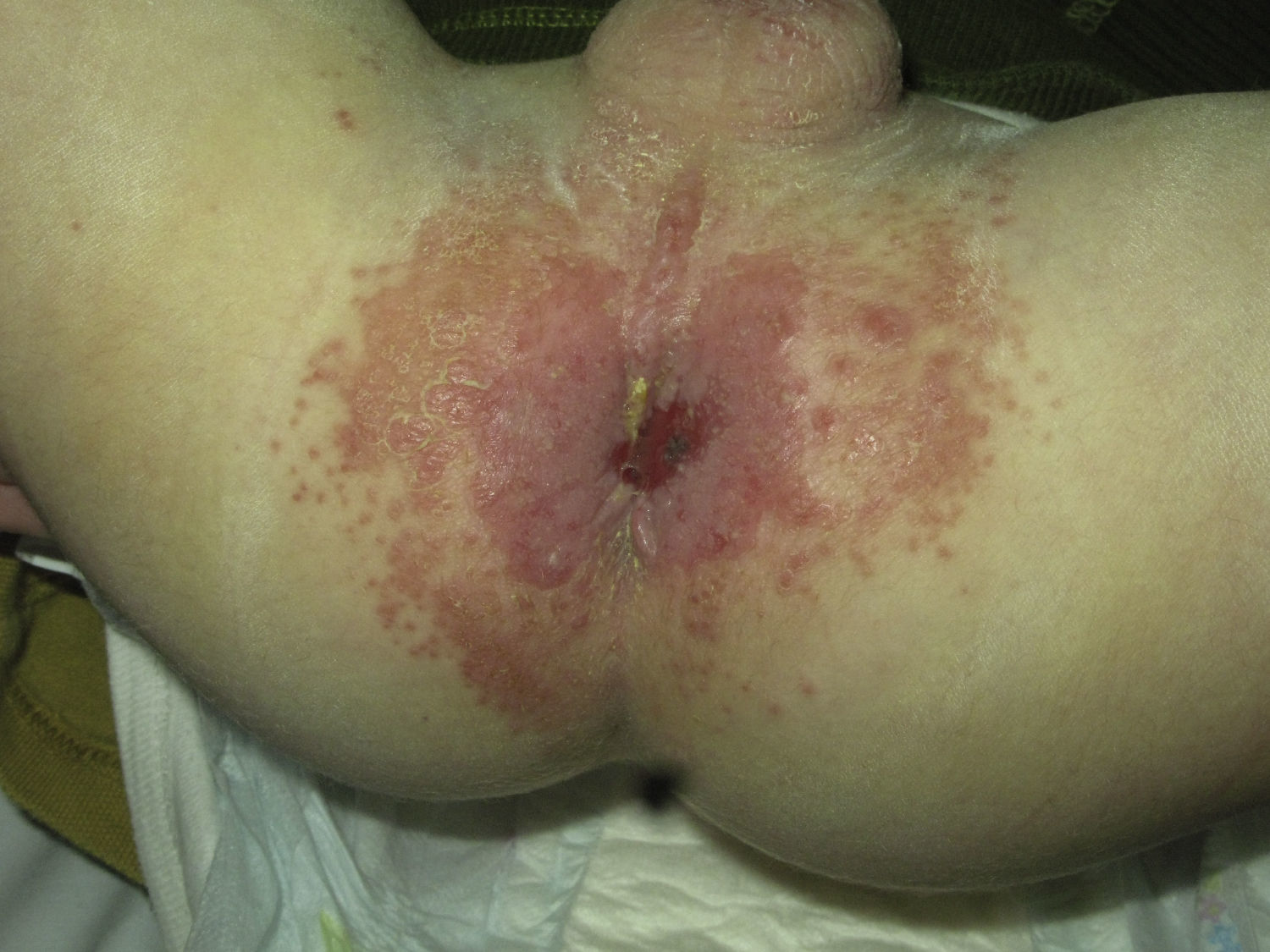
Se afectan los pliegues, especialmente el interglúteo, los submamarios, los axilares y los inguinales. Son áreas con un ambiente óptimo para el desarrollo de la levadura, por un mayor grado de humedad, calor, fricción y oclusión. Se producen fisuras en el fondo de los pliegues y se observan placas eritematosas de bordes bien definidos con un collarete descamativo. Las lesiones «satélite» en forma de pústulas y erosiones son características del intertrigo candidósico (fig. 3). En una serie irlandesa referían dermatitis del pañal recurrente y de difícil tratamiento antes de los 2 años de edad en 5 de 18 pacientes con SPA-133.
- •
Afectación ungueal
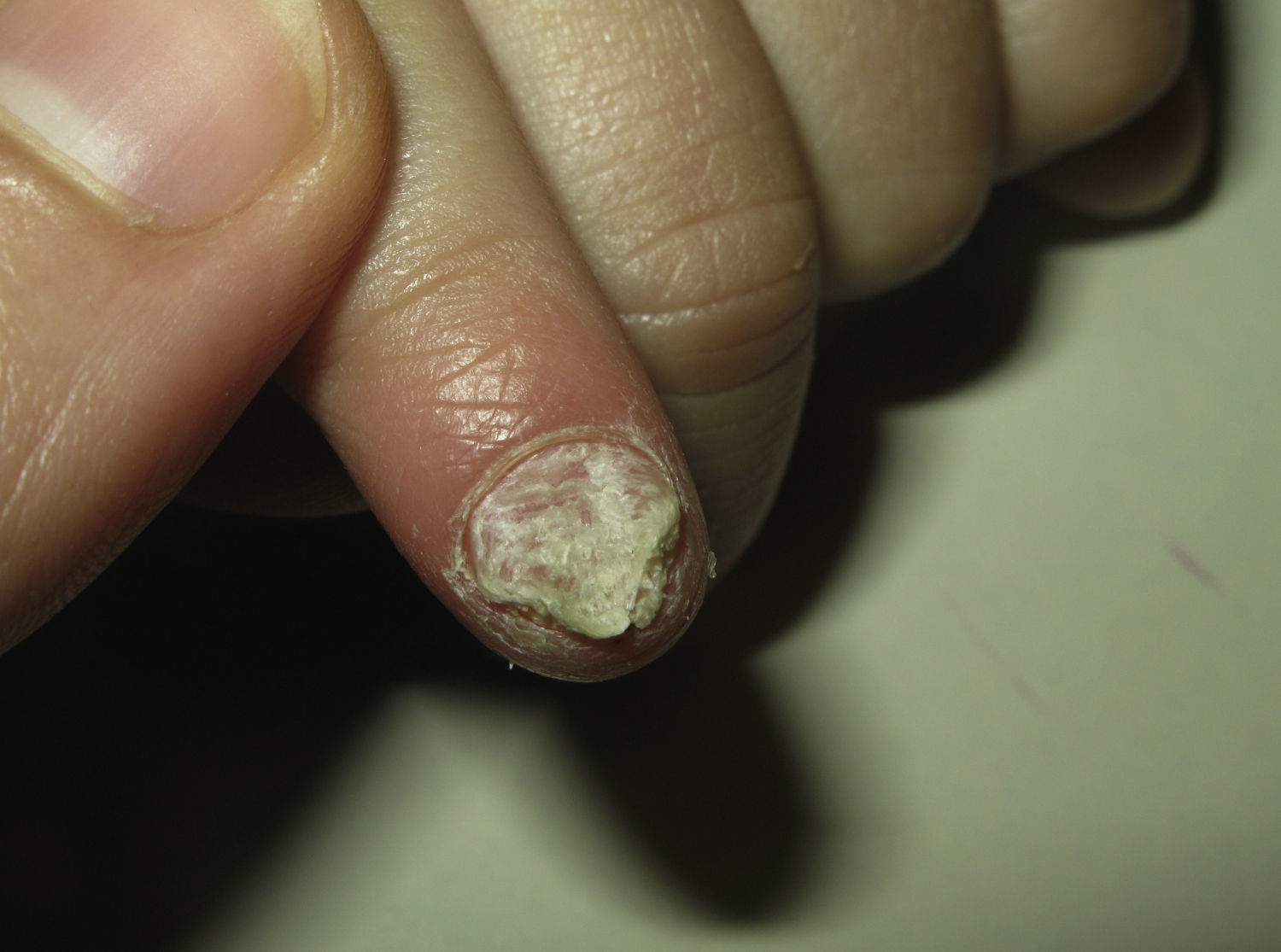
Se produce con mayor frecuencia en situaciones de humedad y maceración. En la paroniquia candidósica se afecta el reborde periungueal en forma de eritema, inflamación y dolor. Si se presiona dicha localización, aparece un exudado purulento bajo el pliegue. Si los síntomas persisten, pueden producirse surcos, decoloración y onicólisis lateral en la tabla ungueal. En algunos casos la tabla ungueal se engrosa y aparece una distrofia ungueal. En una serie de 18 pacientes, el 72% presentaron paroniquia y/onicomicosis. La edad media de aparición eran los 7 años. Las uñas de las manos se afectaban con más frecuencia que las de los pies, siendo las uñas del primer dedo las más afectadas. La tabla ungueal se engrosaba y adquiría una coloración amarillenta. La dificultad terapéutica era la norma en la mayoría de estos pacientes25.
- •
Diagnóstico
El diagnóstico se basa en los síntomas y signos que presenta el paciente y se confirma mediante cultivos. La toma de muestras en las lesiones húmedas de piel y mucosas debe realizarse con hisopos o escobillones estériles provistos de un medio de transporte, ya que la levadura no resiste la desecación y permanece poco tiempo viable en una torunda de algodón. En las lesiones secas, debe practicarse el raspado convencional. El medio de cultivo de las levaduras del género Candida se realiza en el medio glucosado de Sabouraud a una temperatura óptima de 37°.
- •
Tratamiento
En la mayoría de los pacientes con SPA-1 el tratamiento sistémico con fluconazol o itraconazol es casi siempre requerido, prolongado y repetido. Los fármacos tópicos pueden ayudar a controlar más rápidamente los síntomas y a disminuir la tasa de recurrencias34.
- •
Detección y corrección de los factores predisponentes
La infección de las mucosas debe ser controlada y tratada para prevenir el desarrollo de lesiones malignas. Es esencial una estricta higiene oral y una abstención tabáquica y alcohólica. También se recomienda evitar la ingesta de alimentos o productos que dañen la mucosa oral (picantes, ácidos, pastas abrasivas blanqueantes). Han de corregirse las situaciones que favorezcan la humedad, la maceración y la oclusión. En los pacientes portadores de prótesis dentales, es importante que disminuyan su uso y que realicen un correcto lavado y mantenimiento de la misma.
- •
Tratamiento de la candidiasis de piel y mucosas
En las formas cutáneas y en las formas mucosas con afectación leve puede ser suficiente con el tratamiento tópico. Los fármacos poliénicos (nistatina y anfotericina B), los azoles (clotrimazol, miconazol, bifonazol, tioconazol, sertaconazol), las alilaminas (terbinafina y naftifina) y las piridonas (ciclopiroxolamina) son eficaces en las formas leves. Casi todos estos fármacos se encuentran disponibles en forma de cremas, soluciones y óvulos vaginales.
Cuando exista mucositis, se debe iniciar un tratamiento antifúngico tópico. Se recomienda el uso de 2 tipos de derivados poliénicos 4 veces al día. Pueden utilizarse suspensiones de nistatina seguidos de enjuagues de anfotericina B. Esta pauta debe continuarse durante 4-6 semanas y al menos una semana tras la desaparición de los síntomas. La aparición de resistencias es baja con estos agentes, siendo mucho más elevada con los derivados azólicos35.
En casos refractarios al tratamiento tópico o en formas más graves, deben utilizarse antifúngicos sistémicos. Se han obtenido buenos resultados con el uso de fluconazol oral (200-300mg/día en adultos) durante una semana e itraconazol (100mg/día) durante dos semanas.
En pacientes con formas recurrentes, se recomienda que la fase de tratamiento sea continuada por una de profilaxis. Puede utilizarse una suspensión de nistatina 3 veces al día durante una semana al mes. El uso de un enjuague bucal con clorhexidina 2 veces al día durante una semana ejerce un efecto profiláctico similar.
En el caso de que exista una úlcera oral que no cicatriza en un periodo de 4 semanas, será obligatoria la realización de una biopsia.
Las balanitis y vulvovaginitis no complicadas suelen responder bien al tratamiento tópico con cremas y óvulos vaginales de nistatina o derivados imidazólicos. Las cremas se administran 2 veces al día durante un periodo de 7 a 10 días. Los óvulos de clotrimazol (500mg) pueden administrarse como dosis única. Con la terapia oral también se consiguen buenos resultados, se recomienda una dosis única de fluconazol 150mg o itraconazol 100mg/día durante 7 días, o 200mg/día durante 3 días. La pareja debe ser tratada también, aún en ausencia de sintomatología.
- •
Tratamiento de la paroniquia y onicomicosis candidósica
La humedad excesiva debe evitarse en las manos, ya que favorece la infección en la piel y uñas. En los niños, el chupeteo repetido de los dedos favorece la infección. Es importante por tanto modificar los factores predisponentes. Para el tratamiento de la infección se utilizan cremas de polienos o imidazoles de forma prolongada. Hay pocos estudios para valorar la eficacia del fluconazol e itraconazol en esta forma clínica, aunque parece que los resultados son buenos. En cambio, en la onicomicosis hiperqueratósica por Candida la respuesta terapéutica a antifúngicos orales y/o sistémicos es baja. Algunos autores refieren respuestas aceptables con itraconazol oral, aunque se requieren periodos de tratamiento prolongados y un mantenimiento posterior3,8,36. En las formas más erosionadas y atróficas se han conseguido buenas respuestas con laca de amorofilina33.
Como conclusión, en el tratamiento de la CMCC en los pacientes con SPA-1, una vez conseguida la remisión de los síntomas, no debe seguirse una terapia de mantenimiento por el elevado riesgo de aparición de resistencias farmacológicas. Aunque la tendencia al desarrollo de candidiasis oral parece disminuir con el tiempo en algunos pacientes, estos siguen siendo muy susceptibles a tener episodios recurrentes de candidiasis, especialmente tras tratamientos antibióticos prolongados.

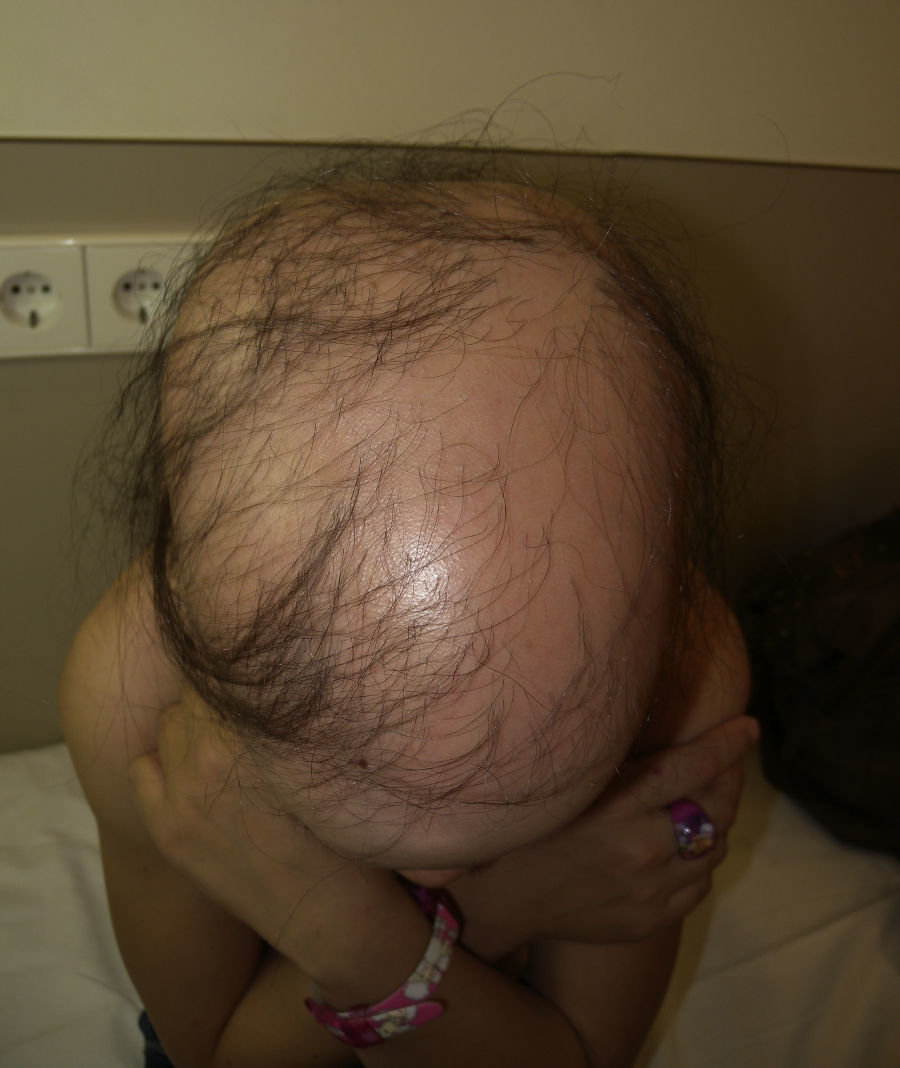
Las manifestaciones cutáneas de base autoinmune están bien descritas en los pacientes con SPA-1. La asociación con alopecia areata se describió por primera vez en 194636. Tras la candidiasis mucocutánea, es la manifestación cutánea más frecuente en los pacientes con SPA-1. Aparece entre los 3 y los 30 años de edad, con una incidencia que oscila entre el 13-40%2,9. Su inicio es más tardío que la de otras anomalías del síndrome; en una serie de 89 pacientes fue la primera manifestación de la enfermedad tan solo en el 4% de los pacientes27. En otra serie de 18 pacientes se desarrolló en algún momento de la evolución en el 33%33. Todos ellos presentaban ya algún otro criterio de la enfermedad y ninguno asociaba un piqueteado o distrofia ungueal. En la mayoría de los casos la alopecia que aparece es en placas, no difiriendo de la alopecia areata aislada, aunque se han descrito también formas universales37 (fig. 4). La mayoría de los pacientes presentan solo un episodio de alopecia y suelen recuperarse por completo. Es una manifestación que aparece tardíamente y es un marcador de cronicidad y mayor gravedad. Algunos pacientes, especialmente mujeres, conciben este componente de la enfermedad como uno de los peores, por la gran afectación psicológica que les produce.

Clínicamente se caracteriza por la aparición de una a varias placas en el cuero cabelludo, aunque pueden afectarse otras localizaciones como cejas, pestañas, vello corporal, axilar y pubiano. Las placas son alopécicas y sin escamas y el signo de Jacqet es positivo (el cuero cabelludo se puede pellizcar). En la periferia se pueden observar los pelos peládicos o en signo de admiración, con el extremo terminal más engrosado. En niños, puede observarse con mayor frecuencia un piquetado ungueal o traquioniquia. Cuando se produce la repoblación, el pelo es fino y apigmentado, adquiriendo posteriormente las características del cabello normal. La evolución y el pronóstico de la alopecia areata son imprevisibles. Se han propuesto dos subtipos de alopecia areata, el «benigno» y el «de mal pronóstico», siendo marcadores de este último la presencia de historia familiar, el inicio precoz de la alopecia y las alteraciones ungueales38,39.
La etiología de la alopecia areata es desconocida, pero parece que existen alteraciones autoinmunes organoespecíficas. Hedstrand et al. encontraron autoanticuerpos contra la tirosina hidroxilasa en el suero de pacientes con alopecia areata40. Esta enzima se expresa en altos niveles en los folículos pilosos. El 44% de los pacientes con SPA-1 presentaron una inmunorreactividad contra la tirosina hidroxilasa expresada in vitro y esta reactividad se correlacionaba con la presencia de alopecia areata (p=0,02).
El tratamiento recomendado es el mismo que en otras formas de alopecia areata aislada38,39.
- •
Inmunosupresores
Se han utilizado corticoides orales en alopecias areatas totales o universales, especialmente en aquellos casos con ANA positivos, con niveles séricos de IgE superiores a 500 UI/L o cuando la relación CD4/CD8 es superior a 4. Los corticoides intralesionales, utilizando acetónido de triamcinolona, se consideran el tratamiento de elección en formas con placas únicas o múltiples que abarquen menos del 50% de la superficie del cuero cabelludo. También se han descrito resultados exitosos con el uso de corticoides tópicos como el propionato de clobetasol al 0,025-0,05%. Como su absorción en el cuero cabelludo es mínima, puede considerarse un tratamiento seguro en niños.
- •
Inmunomoduladores
Se han utilizado múltiples agentes con los que se obtienen resultados dispares. Algunos de los fármacos más utilizados han sido la ciclosporina A, el tacrolimus, la dapsona, la sulfasalazina o la isoprinosina.
- •
Inmunoestimulantes
El objetivo de estos tratamientos es desencadenar un cuadro de «dermatitis de contacto». Dicho proceso inflamatorio podría ser responsable de la remisión de la alopecia areata por un mecanismo aún no conocido por completo. La presencia de linfocitos T supresores podría inhibir la reacción autoinmune y permitir el crecimiento normal del folículo piloso. Entre estos agentes se han utilizado la antralina, en concentraciones de 0,25-1%, con buenos resultados y muy utilizada en niños; el dibutiléster del ácido escuárico y la difenciprona. Este último se considera actualmente el mejor inmunoestimulante y se recomienda en pacientes adultos con alopecias que afectan más del 50% del cuero cabelludo y que no hayan respondido o hayan rechazado la corticoterapia previamente.
- •
Otros tratamientos
El minoxidilo en combinación con otros agentes inmunomoduladores o corticoides tópicos podría mejorar la respuesta terapéutica. Actualmente se recomienda su aplicación al 5% 2 veces al día, media hora antes de la aplicación de un corticoide tópico, tipo propionato de clobetasol al 0,025%.
Se ha implicado a un déficit de zinc en la patogenia de la alopecia areata, aunque dicha teoría no ha podido ser demostrada. Aún así, algunos autores recomiendan la ingesta de aspartato de zinc a una dosis de 50mg dos veces al día.
Es aconsejable también una evaluación global del paciente y proporcionar una adecuada información sobre la evolución y opciones terapéuticas disponibles. Si existe repercusión psicológica, puede ser necesaria la colaboración de los facultativos pertinentes.
Se ha descrito en un tercio de los pacientes en una serie finlandesa de 89 pacientes28. La localización y la extensión son variables, pero la enfermedad puede progresar afectando a gran parte de la superficie corporal. Puede aparecer de forma aislada o en pacientes que presentan también alopecia areata.
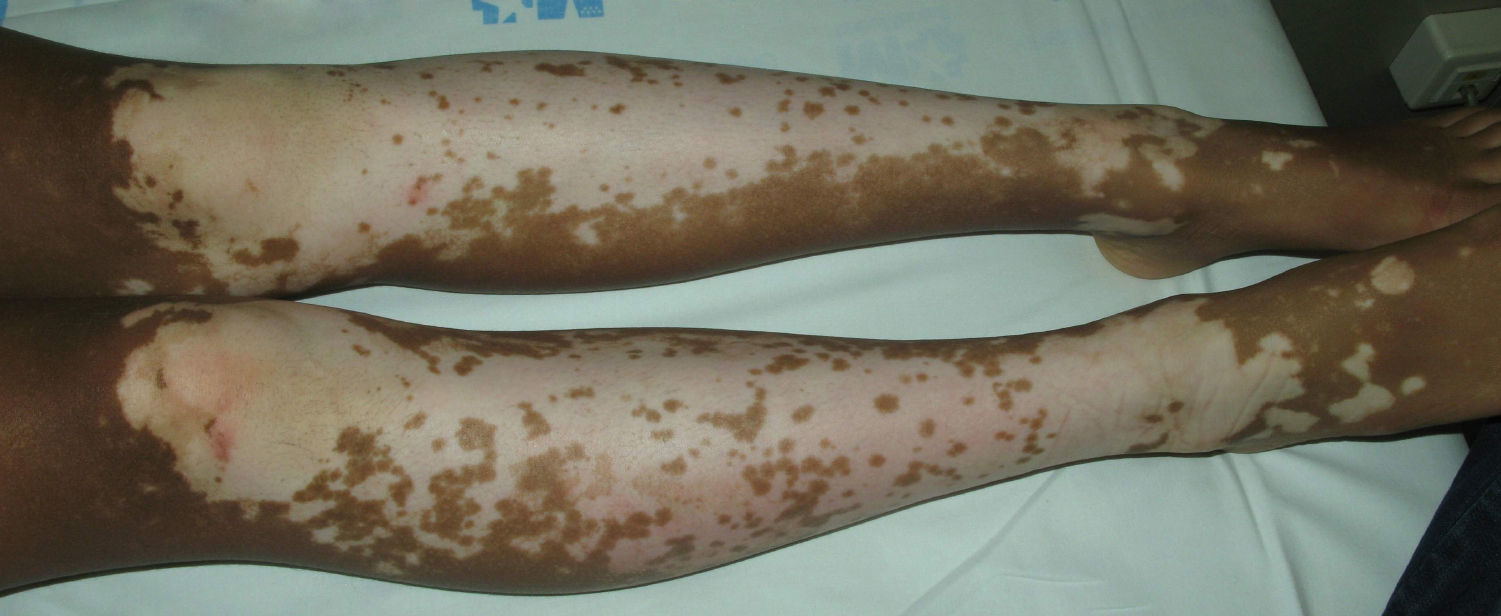
El vitíligo se caracteriza por la aparición progresiva de manchas acrómicas de color blanco «lechoso», de tamaños muy variables y de contornos geográficos e irregulares (fig. 5). Las lesiones suelen tener localización distal y periorificial, a menudo son simétricas o zoniformes, progresivas y pueden repigmentarse espontánea y parcialmente. Existe una disminución o ausencia de melanocitos y melanina en las zonas afectadas. La hipótesis inmunológica sugiere un trastorno autoinmunitario, con destrucción y disfunción de los melanocitos, que en algún momento se volvieron antigénicos. Esta hipótesis se apoya en el hallazgo de anticuerpos específicos circulantes. Se han descrito también casos de halo nevus y de poliosis33.

Se han encontrado autoanticuerpos fijadores del complemento contra los melanocitos en pacientes con SPA-1 y CMCC, pero no en pacientes con vitíligo aislado o asociado a otras manifestaciones autoinmunes41-43. En estos pacientes también se han encontrado anticuerpos contra la L-aminoácido aromático decarboxilasa44. No se conoce aún el papel de dichos anticuerpos. Hedstrand et al. encontraron reactividad contra el factor de transcripción SOX10 en la mayoría de los pacientes con SPA-1 y vitíligo, pero también en un subgrupo de pacientes con vitíligo idiopático. Por ello SOX10 podría tener un papel relevante en el desarrollo de esta enfermedad45.
El diagnóstico del vitíligo es fundamentalmente clínico, por las características morfológicas, curso y distribución de las lesiones. Por la afectación de áreas muy visibles, puede tener una importante repercusión psicosocial. Existe además una sensibilidad aumentada de la piel acrómica a la radiación solar. Es por ello importante informar a los pacientes sobre la naturaleza de la enfermedad, los tratamientos disponibles y las expectativas reales que deben formarse. Se recomienda especial precaución con la exposición solar por el mayor riesgo de quemadura solar.
Como en otras formas de vitíligo aislado, pueden utilizarse diferentes tratamientos, entre los que se incluyen la fototerapia, la corticoterapia tópica, los inmunomoduladores tópicos como el tacrolimus o el pimecrolimus, o despigmentantes en las formas más avanzadas. Actualmente en los casos con afectación moderada y extensa la radiación con UVB de banda estrecha 2-3 veces por semana se considera el tratamiento de elección46. Es un tratamiento eficaz y sin efectos secundarios relevantes, aunque como no se conocen los efectos adversos a largo plazo, no se deben superar las 200 sesiones. Otras opciones terapéuticas incluyen el tratamiento quirúrgico con injertos de piel autólogos, aunque estas técnicas aún no están consolidadas y deben perfeccionarse.
Otras manifestaciones cutáneasSe ha documentado un exantema con fiebre en algunos pacientes con SPA-1, describiéndose formas más graves en los pacientes más jóvenes27. En la serie finlandesa, 13 pacientes presentaron exantemas morbiliformes con fiebre. Algunos eran de duración fugaz y en la mayoría de los casos ocurrían antes de los 5 años de edad. Aparecieron nuevos episodios durante un periodo de 2 a 14 meses. En 2 pacientes biopsiados, la histología mostró una vasculitis de predominio linfoplasmocitario2.
Alteraciones ectodérmicasEn la descripción del SPA-1 se incluyen anomalías de las estructuras derivadas del ectodermo, que incluyen una hipoplasia del esmalte dental, distrofia y piqueteado ungueal y una calcificación de la membrana timpánica47. Los síndromes de displasias ectodérmicas se caracterizan por dos o más anomalías en estructuras derivadas del ectodermo: pelo, dientes, uñas, glándulas sudoríparas y otras. Además, suelen asociar manifestaciones en otros órganos48. Las mutaciones en el gen AIRE responsables del SPA-1 no guardan relación con las estructuras ectodérmicas. Los datos recogidos en los estudios más recientes apuntan que parece poco probable que este síndrome sea una displasia ectodérmica primaria33.
En varias series se ha reportado una distrofia ungueal en muchos de los pacientes, pero no se especifica si la exploración física fue realizada por un dermatólogo o por un facultativo especializado en patología ungueal8,9,27,33. Cuando se observen alteraciones ungueales en estos pacientes, deben realizarse cultivos de las lesiones (fig. 6). En una serie de 50 pacientes9, 13 presentaban un piquetado ungueal en ausencia de infección candidósica. Ninguno tenía distrofia ungueal. Tampoco se documenta que presentaran alopecia areata u otras causas que justificaran el piqueteado ungueal. Los autores concluyen que la diferenciación entre una onicomicosis candidósica y una verdadera distrofia ungueal es complicada.

En una serie de 20 pacientes noruegos, 2 tenían distrofia ungueal8. Ambos presentaban signos clínicos típicos y cultivo positivo para Candida. En esta misma serie 8 pacientes fueron diagnosticados de alopecia areata, y ninguno de ellos mostraba alteraciones ungueales, ya sea en forma de piqueteado o de distrofia ungueal. Por todo ello, algunos autores dudan de que en el SPA-1 las anomalías del ectodermo observadas sean otra parte más del espectro de la enfermedad y las consideran alteraciones secundarias a la infección candidósica crónica33.
Entre las anomalías dentales, se ha incluido una hipoplasia del esmalte dental49,50. Este hallazgo estuvo presente en el 75% de los pacientes de la serie finlandesa27. Es probable que los defectos dentales, al igual que los ungueales, sean secundarios a la infección oral recurrente, a la malnutrición y a la hipocalcemia. Parece poco probable que se trate de un fallo primario en la morfogénesis33.
Manifestaciones ocularesSe han descrito queratitis, ojo seco, cataratas, iridociclitis, desprendimiento de retina y atrofia óptica51,52. La queratitis es la alteración ocular más frecuente y aparece en el 20-25% de los pacientes27. Si no se trata adecuadamente, puede conducir a la ceguera. Suele ser una manifestación temprana de la enfermedad y se presenta como fotofobia, blefaroespasmo y disminución del lagrimeo. Parece que se trata de un proceso autoinmune contra el epitelio corneal. Como tratamiento se han utilizado corticoides y ciclosporina A tópicos. Es importante también prevenir las infecciones oculares y tratarlas cuando se produzcan. El uso de vitamina A tópica podría prevenir la aparición de úlceras corneales53.
DiagnósticoEl diagnóstico del SPA-1 no debe pasar desapercibido, porque un diagnóstico precoz permite un tratamiento sustitutivo en las fases en las que aún no existen trastornos relevantes. Por otra parte, no hay que precipitarse en la confirmación del síndrome si solo se detectan anticuerpos sin otras alteraciones hidroelectrolíticas o clínicas. La detección de los mismos no es un marcador fiable de enfermedad. La actitud recomendada es realizar un control regular de los marcadores hormonales e inmunológicos en todos los pacientes con una endocrinopatía inmunitaria, para diagnosticar precozmente un SPA1,54.
Cuando se diagnostique una enfermedad endocrinológica glandular, se debe descartar la existencia de otros procesos autoinmunes y realizar un cribado con los autoanticuepos de cada enfermedad. La presencia de autoanticuerpos no significa que exista enfermedad o que esta se vaya a producir. Algunos autoanticuerpos fluctúan y pueden desaparecer espontáneamente sin producir alteraciones funcionales. Esto traduce una agresión inmunológica transitoria. Para evaluar a los portadores de autoanticuerpos positivos, se deben determinar periódicamente los niveles de la hormona estimuladora del tiroides, la calcemia y fosfatemia, las glucemias basales y la PTH. De esta forma, podemos establecer el diagnóstico definitivo de SPA-1 en una fase preclínica. De forma general, en los pacientes con una endocrinopatía autoinmune uniglandular está indicado realizar un cribado funcional para SPA cada 3 años hasta los 75 años de edad54. Si se descubre una segunda enfermedad endocrinológica autoinmune, se debe continuar con una medición de los autoanticuerpos específicos de órgano. También sería recomendable realizar un cribado funcional de endocrinopatías autoinmunes en los familiares de primer grado en aquellos pacientes con un diagnóstico reciente de SPA-1. Actualmente está indicado el estudio genético para confirmar definitivamente el diagnóstico si se encuentra la mutación en el gen AIRE10,11.
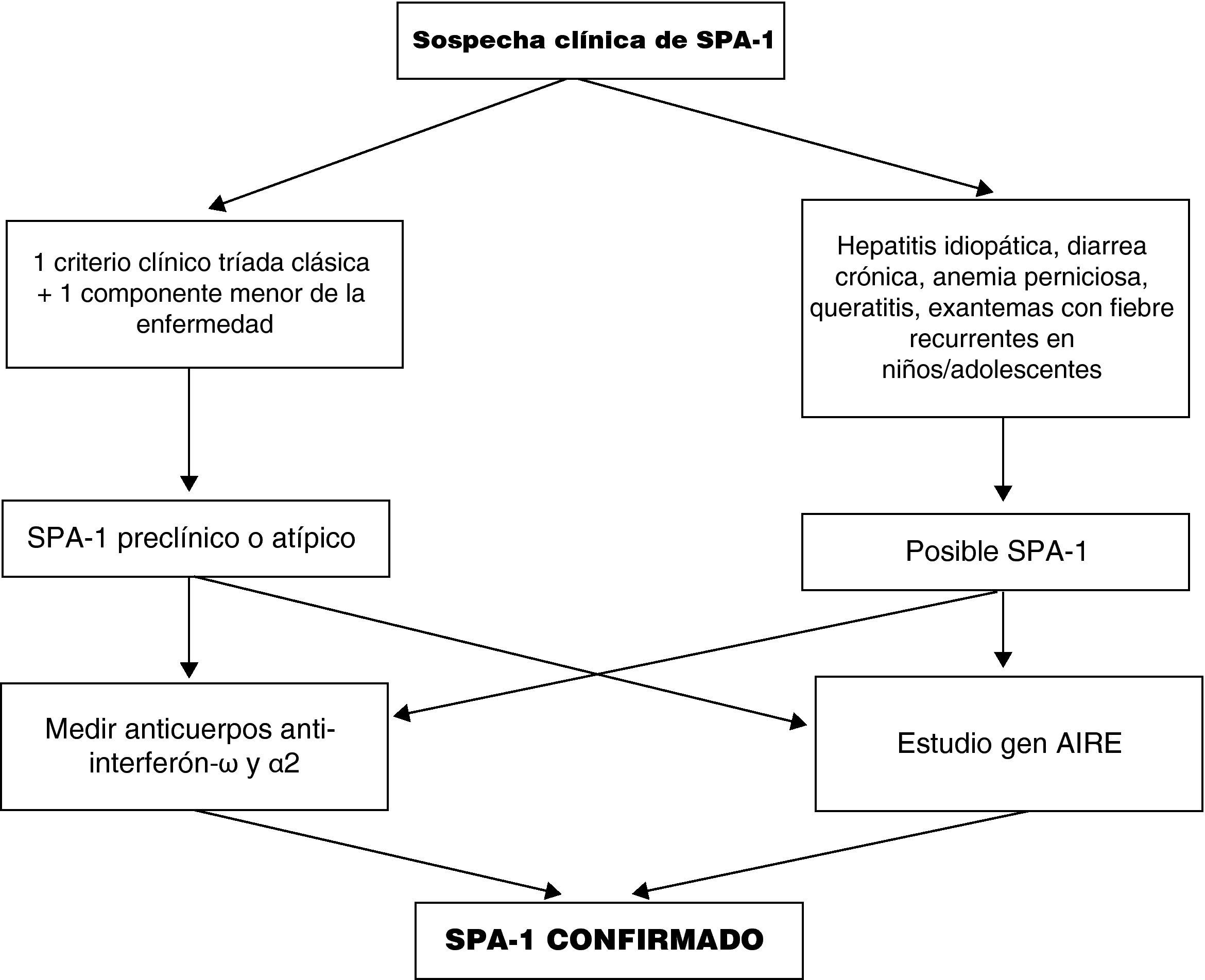
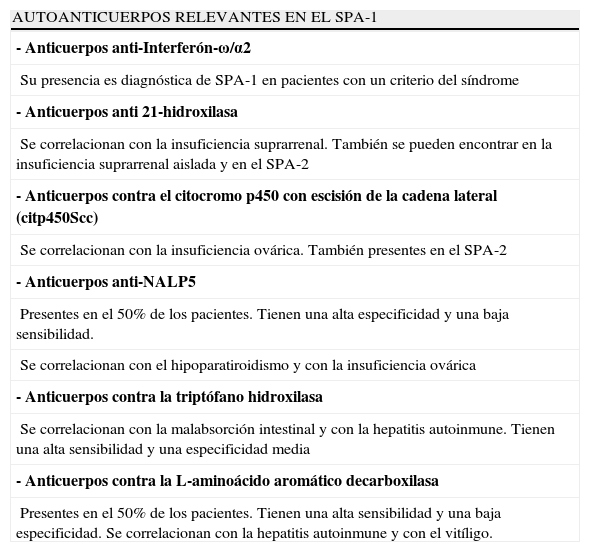
Los avances en genética y en el reconocimiento de las moléculas implicadas en la patogenia de los síndromes autoinmunes han facilitado un diagnóstico más temprano en este grupo de enfermedades. El diagnóstico basado en los criterios clínicos dejaba a muchos pacientes sin reconocer y en un potencial «peligro». En un 10-15% de los pacientes las primeras manifestaciones clínicas de la enfermedad no incluyen ninguno de los 3 criterios diagnósticos clásicos25,27. Por ello, en pacientes con alta sospecha de SPA, la medición de los niveles de anticuerpos contra el interferón-ω y α2 puede confirmar el diagnóstico, según algunos estudios recientes55-57, siempre y cuando se haya excluido la presencia de un timoma y miastenia gravis. Si no se pueden medir estos anticuerpos, la presencia de anticuerpos contra las enzimas 21 hidroxilasa, la L-aminoácido aromático decarboxilasa y la triptófano hidroxilasa también apoyan el diagnóstico de SPA-1 (tabla 2). Finalmente el diagnóstico puede confirmarse en el laboratorio al encontrarse mutaciones en el gen AIRE en más del 95% de los casos. Esta técnica es costosa y un resultado negativo no excluye el diagnóstico. Aún son necesarias nuevas tecnologías que mejoren las pruebas diagnósticas genéticas (fig. 7).
Autoanticuerpos en los pacientes con SPA-1
| AUTOANTICUERPOS RELEVANTES EN EL SPA-1 |
| - Anticuerpos anti-Interferón-ω/α2 |
| Su presencia es diagnóstica de SPA-1 en pacientes con un criterio del síndrome |
| - Anticuerpos anti 21-hidroxilasa |
| Se correlacionan con la insuficiencia suprarrenal. También se pueden encontrar en la insuficiencia suprarrenal aislada y en el SPA-2 |
| - Anticuerpos contra el citocromo p450 con escisión de la cadena lateral (citp450Scc) |
| Se correlacionan con la insuficiencia ovárica. También presentes en el SPA-2 |
| - Anticuerpos anti-NALP5 |
| Presentes en el 50% de los pacientes. Tienen una alta especificidad y una baja sensibilidad. |
| Se correlacionan con el hipoparatiroidismo y con la insuficiencia ovárica |
| - Anticuerpos contra la triptófano hidroxilasa |
| Se correlacionan con la malabsorción intestinal y con la hepatitis autoinmune. Tienen una alta sensibilidad y una especificidad media |
| - Anticuerpos contra la L-aminoácido aromático decarboxilasa |
| Presentes en el 50% de los pacientes. Tienen una alta sensibilidad y una baja especificidad. Se correlacionan con la hepatitis autoinmune y con el vitíligo. |

La morbimortalidad está incrementada respecto a la población general, pero el curso de la enfermedad difiere en cada paciente25,27,58. El espectro es amplio. Hay casos en los que la enfermedad se mantiene estable con un hipoparatiroidismo controlado y otros en los que se afectan la mayoría de las glándulas endocrinas y asocian otras manifestaciones no endocrinológicas, como hepatitis autoinmune, gastritis atrófica, vitíligo o alopecia areata, con una importante repercusión psicológica. El pronóstico está condicionado también por el cumplimiento terapéutico del paciente. En la serie finlandesa, la mortalidad era mayor en pacientes incumplidores por aislamiento social, alcoholismo o comorbilidad psiquiátrica27. La siguiente causa de mortalidad era por carcinomas epidermoides orales o esofágicos. Otros procesos que incrementan la mortalidad son un fallo suprarrenal agudo, una hepatitis autoinmune fulminante, un fracaso renal agudo o una bronquiolitis5,59. La mortalidad reportada ocurría en la mayoría de los casos en pacientes mayores de 60 años y podría reducirse con un adecuado seguimiento y tratamiento25.
Abordaje y manejo de los pacientes con SPA-1El objetivo es que los pacientes puedan desarrollar una vida normal y evitar situaciones que puedan desencadenar crisis endocrinas. Deben conocer potenciales manifestaciones del síndrome para consultar de forma precoz. Se requiere un manejo multidisciplinar, que incluya la atención por parte de endocrinos, dermatólogos, digestivos y psiquiatras. Si aparecen otras manifestaciones, el paciente deberá ser derivado al especialista correspondiente. Se recomienda también la vacunación contra Pneumococcus pneumoniae, por el riesgo de desarrollar hipo/asplenia que presentan estos pacientes25. Han de conocer el protocolo de tratamiento que deben seguir en situaciones que puedan desequilibrar su patología, tales como infecciones, cirugías u otras situaciones estresantes.
El SPA-1 se considera un modelo de enfermedad autoinmune. Se están realizando múltiples estudios para la identificación de los genes implicados en la autotolerancia inmunológica. Se buscan nuevas terapias, especialmente inmunomoduladoras, que puedan frenar o revertir las alteraciones autoinmunes60,61. La terapia génica supone otra alternativa en desarrollo para el tratamiento de estos pacientes.
- •
En el SPA-1 se produce una destrucción progresiva de los tejidos glandulares endocrinos que puede desencadenar manifestaciones variables que incluyen hipoparatiroidismo, insuficiencia suprarrenal, hipotiroidismo, hipogonadismo o diabetes mellitus insulinodependiente.
- •
Además de la afectación endocrina, se han descrito otras alteraciones autoinmunitarias no endocrinológicas (hepatitis autoinmune, gastritis atrófica, malabsorción intestinal) y anomalías ectodérmicas (hipoplasia del esmalte dentario, queratitis, distrofia ungueal).
- •
Para el diagnóstico del SPA-1 se han utilizado criterios clínicos. La identificación del gen AIRE como responsable de la enfermedad ha servido para establecer el diagnóstico definitivo y detectar nuevos casos precozmente antes de que la enfermedad se manifieste de forma florida.
- •
La tríada clásica del SPA-1 incluye la CMCC, el hipoparatiroidismo y la insuficiencia suprarrenal. Estos tres componentes aparecen en un preciso orden cronológico, pero los tres, de forma conjunta, solo están presentes en el 35-50% de los pacientes.
- •
Muchos de los pacientes con SPA-1 desarrollarán patología cutánea, entre las que se incluyen como más frecuentes una CMCC, alopecia areata, vitíligo, exantemas febriles y distrofia ungueal.
- •
Los pacientes con CMCC deben ser evaluados periódicamente para reconocer precozmente los síntomas de una hipofunción glandular asociada.
- •
La morbimortalidad en los pacientes con SPA-1 está incrementada respecto a la población general, por lo que es importante un manejo multidisciplinar por parte de diferentes facultativos para que estos pacientes puedan desarrollar una vida normal y evitar situaciones que puedan desencadenar «crisis» endocrinas.