




Global developmental delay (GDD) and intellectual disability (ID) are common reasons for consultation in paediatric neurology. Results from aetiological evaluations of children with GDD/ID vary greatly, and consequently, there is no universal consensus regarding which studies should be performed.
Material and methodWe review our experience with determining aetiological diagnoses for children with GDD/ID who were monitored by the paediatric neurology unit over the 5-year period between 2006 and 2010.
ResultsDuring the study period, 995 children with GDD/ID were monitored. An aetiological diagnosis was established for 309 patients (31%), but not in 686 (69%), despite completing numerous tests. A genetic cause was identified in 142 cases (46% of the total aetiologies established), broken down as 118 cases of genetic encephalopathy and 24 of metabolic hereditary diseases. Our data seem to indicate that diagnosis is easier when GDD/ID is associated with cerebral palsy, epilepsy, infantile spasms/West syndrome, or visual deficit, but more difficult in cases of autism spectrum disorders. Genetic studies provide an increasing number of aetiological diagnoses, and they are also becoming the first step in diagnostic studies. Array CGH (microarray-based comparative genomic hybridisation) is the genetic test with the highest diagnostic yield in children with unexplained GDD/ID.
DiscussionThe cost-effectiveness of complementary studies seems to be low if there are no clinically suspected entities. However, even in the absence of treatment, aetiological diagnosis is always important in order to provide genetic counselling and possible prenatal diagnosis, resolve family (and doctors’) queries, and halt further diagnostic studies.
El retraso global del desarrollo (RGD) y la discapacidad intelectual (DI) son motivos de consulta frecuentes en la práctica neuropediátrica. El rendimiento de los estudios diagnósticos en niños con RGD/DI varía ampliamente y, en consecuencia, no hay acuerdo universal respecto a los estudios que se deben realizar.
Material y métodoRevisamos nuestra experiencia en el diagnóstico etiológico de los niños con RGD/DI valorados en la consulta de Neuropediatría durante un periodo de 5 años: 2006-2010.
ResultadosDurante el periodo de estudio fueron valorados 995 niños con RGD/DI. El diagnóstico etiológico fue establecido en 309 (31%) y no en 686 (69%), a pesar de múltiples estudios realizados. En 142 niños, el 46% de los casos con diagnóstico etiológico establecido, la causa es genética: 118 encefalopatías genéticas y 24 enfermedades metabólicas hereditarias. Nuestros datos indican que establecer un diagnóstico etiológico es más fácil cuando el RGD/DI está asociado a parálisis cerebral infantil, epilepsia, espasmos infantiles/síndrome de West o déficit visual, pero más difícil en casos de trastorno del espectro autista. Los estudios genéticos están incrementando los diagnósticos etiológicos y constituyéndose en el primer escalón de estudio. El microarray comparative genomic hybridisation es la prueba con mayor rentabilidad diagnóstica en el estudio de RGD/DI.
DiscusiónEl coste-efectividad de los exámenes complementarios es aparentemente bajo en ausencia de orientación clínica. Incluso en ausencia de tratamiento, el diagnóstico etiológico es importante para establecer un consejo genético y posible diagnóstico prenatal, resolver cuestiones a padres y profesionales, y cesar la realización de más pruebas complementarias.
Global psychomotor delay (GDD) and intellectual disability (ID) are frequent reasons for visiting a paediatric neurologist. Estimated prevalence of psychomotor and mental retardation oscillates between 1% and 10%.1 There is no established aetiological diagnosis in 50% to 80% of all cases.2 Yield of diagnostic evaluation for children with GDD/ID varies greatly (10%–81%), which reflects many factors including differences between study populations and durations. This situation makes systematic reviews more complicated to perform.3 Consequently, there is no consensus regarding which studies should be performed on children with GDD/ID.1–11 Studies should also be updated and adapted to dynamic social, technical and scientific developments. This article provides a retrospective review of the experience with aetiological diagnosis of GDD/ID cases in our hospital's paediatric neurology unit over the 5-year period between 2006 and 2010.
Material and methodsThe paediatric neurology unit at our tertiary hospital was established in May 1990 and its database contains all the cases evaluated by the unit since that date. This database is updated with all new events related to symptoms, progression, treatment, or complementary studies.12–14
In 2008, we expanded our diagnostic protocol for different paediatric neurological diseases, such as prenatal encephalopathies or GDD/ID, with a view to identifying genetic alterations and inborn errors of metabolism (IEM). We applied this diagnostic protocol to new cases as well as to older cases without an aetiological diagnosis. Extensive urine and blood biochemistry studies, neuroimaging studies (mostly brain MRI scans), karyotyping and fragile-X syndrome testing, and other specific genetic studies had been completed for most of the children included in our review. Those studies are listed in the ‘Results’ section. We established a diagnosis of GDD for those patients younger than 5 years who presented altered neurodevelopment in 2 or more categories of the Denver Developmental Screening Test or Haizea-Llevant scale. A diagnosis of ID was established for patients older than 5 years who presented an intelligence quotient of <85 or needed special education or significant curriculum adaptation.
We provide a retrospective review of our experience with aetiological diagnosis of GDD/ID cases during the 5-year period between 2006 and 2010.
Our study analyses case incidence during this period and the prevalence of those previously diagnosed cases that were followed up during the study period. We included both children with isolated GDD/ID and those with GDD/ID associated with other concomitant diseases such as childhood cerebral palsy (CCP), autism spectrum disorders (ASD), or epilepsy; cases of regression are also included. ASD cases displaying normal intelligence (high-functioning autism) were excluded. We reviewed the diagnosis and the studies performed for patients with no established aetiological diagnosis.
Pearson's chi-square test with one degree of freedom was used to establish the statistical relationship between different concomitant disorders and having GDD/ID or not.
ResultsBetween 1 January 2006 and 31 December 2010, 6108 children were added to the database. A total of 995 (16.5%) presented GDD/ID.
Despite completion of multiple studies, no aetiological diagnoses were assigned in 686 cases (69%). Aetiological diagnoses were provided in 309 cases (31%) (Table 1).
Patients with an aetiological diagnosis for global developmental delay and intellectual disability (n=309).
| Diseases | N |
| Prenatal encephalopathies of disruptive origin | 41 (13%) |
| Congenital infections (13 CMV, 3 toxoplasmosis) | 19 |
| Teratogen-related encephalopathies (16 FAS) | 18 |
| Encephalopathy related to vascular disruptive syndrome of monozygotic twins | 4 |
| Genetic encephalopathies | 118 (38%) |
| Down syndrome | 12 |
| Fragile-X syndrome | 7 |
| Subtelomeric deletions | 7 |
| Patau syndrome | 1 |
| Other chromosomal defects diagnosed by CGH arraya | 21 |
| Tuberous sclerosis | 11 |
| Congenital myotonic dystrophy | 4 |
| Rett syndrome (4 MECP2 and 2 CDKL5 mutations) | 6 |
| Dravet spectrum disorder with SCN1A mutation | 9 |
| Prader–Willi syndrome | 5 |
| Angelman syndrome | 4 |
| Neurofibromatosis type 1 | 5 |
| Other genetic encephalopathiesb | 16 |
| Inborn errors of metabolism | 24 (7.8%) |
| Mitochondrial diseases | 3 |
| Lysosomal diseasesc | 7 |
| Disorders of intermediary metabolismd | 6 |
| X-ALDe | 3 |
| Other IEMsf | 5 |
| Perinatal encephalopathies | 102 (33%) |
| Post-natal encephalopathiesg | 20 (6.55%) |
| Brain tumours | 4 (1.3%) |
| Hemispheric tumour | 2 |
| Cerebellar tumour | 1 |
| Midline tumour | 1 |
CGH array: microarray-based comparative genomic hybridisation; CMV: cytomegalovirus; FAS: fetal alcohol syndrome; X-ALD: X-linked adrenoleukodystrophy.
Other genetic encephalopathies: 3 cases of lissencephaly with LIS1 mutation, 3 cases of dystrophinopathy, 2 with Williams–Beuren syndrome, 2 with Joubert syndrome, 1 with CRASH syndrome and L1CAM mutation, 1 with syntelencephaly and ZIC2 deletion, 1 with Proteus syndrome, 1 with Waardenburg syndrome, 1 with Coffin–Siris syndrome, and 1 with Cornelia de Lange syndrome.
Lysosomal diseases: one case each of Hurler syndrome, neuronal ceroid-lipofuscinosis type 2 (NCL2), and Krabbe disease; 4 cases of metachromic leukodystrophy.
Six disorders of intermediary metabolism: 2 siblings with 4-hydroxybutyric aciduria, 1 case each of homocystinuria, nonketotic hyperglycinemia, maple syrup urine disease, and biotinidase deficiency.
Among cases in which an aetiological diagnosis was established, 142 (46%) had a genetic cause: 118 genetic encephalopathies and 24 IEM. In 126 cases, diagnosis was based on medical history (102 perinatal encephalopathies, 20 post-natal encephalopathies, and 4 brain tumours). Cases diagnosed using the medical history were excluded from the analytical study. As a result, a total of 869 GDD/ID cases were analysed (686 without an established diagnosis and 183 diagnosed).
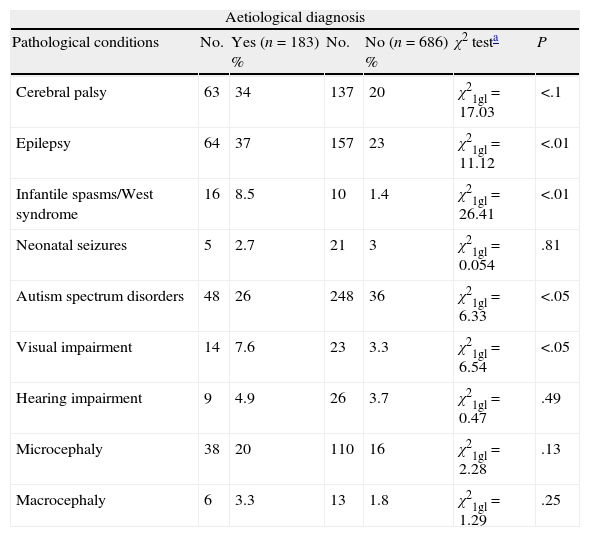
We have analysed the relationship between obtaining an aetiological diagnosis and the association with specific diseases such as CCP, epilepsy, infantile spasms/West syndrome, microcephaly, macrocephaly, neonatal seizures, ASD, visual impairment, or hypoacusia in all cases with GDD/ID. We excluded cases (n=126) diagnosed based on clinical history. Our data show a statistically significant association between obtaining an aetiological diagnosis and presence of CCP, epilepsy, infantile spasms/West syndrome, or visual impairment. There is no statistically significant association between ASD and presenting an aetiological diagnosis (Table 2).
Aetiological diagnosis of patients with GDD/ID and other disorders, and the association between identifying the aetiological diagnosis and the disorder in question. Cases due to prenatal and perinatal encephalopathies and brain tumours have been excluded.
| Aetiological diagnosis | ||||||
| Pathological conditions | No. | Yes (n=183) % | No. | No (n=686) % | χ2 testa | P |
| Cerebral palsy | 63 | 34 | 137 | 20 | χ21gl=17.03 | <.1 |
| Epilepsy | 64 | 37 | 157 | 23 | χ21gl=11.12 | <.01 |
| Infantile spasms/West syndrome | 16 | 8.5 | 10 | 1.4 | χ21gl=26.41 | <.01 |
| Neonatal seizures | 5 | 2.7 | 21 | 3 | χ21gl=0.054 | .81 |
| Autism spectrum disorders | 48 | 26 | 248 | 36 | χ21gl=6.33 | <.05 |
| Visual impairment | 14 | 7.6 | 23 | 3.3 | χ21gl=6.54 | <.05 |
| Hearing impairment | 9 | 4.9 | 26 | 3.7 | χ21gl=0.47 | .49 |
| Microcephaly | 38 | 20 | 110 | 16 | χ21gl=2.28 | .13 |
| Macrocephaly | 6 | 3.3 | 13 | 1.8 | χ21gl=1.29 | .25 |
No., number of cases with a specific condition; GDD/ID, global developmental delay and intellectual disability; P, statistically significant association between presenting a specific condition and the possibility of assigning an aetiological diagnosis; %, percentage of patients displaying a specific condition with and without an aetiological diagnosis.
The following complementary tests were performed for the GDD/ID group without an established diagnosis.
Normal biochemistry tests: 550 ammonia tests, 518 amino acids tests, 525 lactic acid tests, 497 CK tests, 448 thyroid hormone tests, 340 long chain fatty acid analyses, 282 o-tolidine tests, 298 homocysteine tests, 288 ceruloplasmin tests, 281 copper tests, 250 organic acid tests, 179 CDT tests (detection of congenital disorder of glycosylation [CDG]) and 13 neurotransmitter studies in cerebrospinal fluid.
Normal genetic tests: 457 karyotyping studies, 399 fragile-X screenings, 59 subtelomeric deletion studies, 50 CGH arrays, 46 Angelman syndrome tests, 27 Prader–Willi syndrome tests, 18 myotonic dystrophy tests, and mutation tests (12 for MECP2, 7 for CDKL5, 16 for SCN1A and GABRG2). We performed a retrospective analysis for cytomegalovirus DNA in dried blood spots from newborn screening tests in 9 cases.
Results from 284 brain MRI scans, 141 head CT scans, and 69 MR spectroscopy scans were also normal.
Some neuroimaging studies yielded abnormal results but were not sufficient to establish an aetiological diagnosis: 107 MRI scans and 77 head CT scans.
DiscussionOur series seems to indicate that assigning an aetiological diagnosis is easier when GDD/ID is associated with CCP, infantile spasms/West syndrome or visual impairment, but more difficult in cases of ASD (Table 2).
Obviously, some entities are easy to diagnose based on clinical signs, such as Down syndrome, tuberous sclerosis, or neurofibromatosis type 1; this increases the genetic yield of complementary studies, especially genetic tests.
We did not analyse the presence of dysmorphic features or skin anomalies, factors which increase the probability of delivering aetiological diagnoses.
While it was not possible to link the degree of ID to the likelihood of determining an aetiological diagnosis, such a diagnosis is more difficult to assign in cases of mild disability.
IEM screening in children with GDD/ID has a diagnostic yield of between 0.2% and 4.6%, depending on the presence of clinical indicators and the range of the studies performed. CDT testing (percentage of carbohydrate-deficient transferrin) to diagnose CDG syndromes has a diagnostic yield of up to 1.4%, and tests for creatine synthesis and transport defects has a yield of up to 2.8%.8 In our series, we did not perform urine studies for creatine synthesis and transport defects; although these defects can also be detected by MR spectroscopy, routine testing should screen for them given that these conditions are treatable.
Non-specific biochemistry studies have a very low diagnostic yield and they are not very useful in children older than 2–3 years with isolated GDD/ID.
Genetic encephalopathies, congenital infections, peroxisomal or mitochondrial or lysosomal disorders, CDG syndromes, and other IEMs can display indistinguishable clinical signs in their early stages. IEMs are a rare cause of GDD/ID (approximately 1%), especially if there are no signs or symptoms indicating a metabolic problem. However, identifying an IEM can be crucial to the patient's outcome. It is possible that studying relatively rare IEMs would have a greater impact on families and the society than studying genetic syndromes, considering that the former can affect treatment and outcome. The possibility of starting effective treatment should have a more significant effect on clinical practice than the figures for diagnostic yield.3,8
There is no general consensus on performing neuroimaging studies when evaluating a child with GDD/ID. Recommendations vary from performing neuroimaging studies on all patients to limiting their use to cases in which scans are clinically indicated.3 Except in some emergency situations, the current test of choice for studying encephalopathies of all types is MRI. Neuroimaging can establish the aetiological diagnosis, as in the 2 cases of Joubert syndrome we present and in some cases of tuberous sclerosis. Although it does not determine the diagnosis in some patients, it can help identify causes in specific cases, just as a dysmorphology examination can orient doctors towards a clinical diagnosis.3 MRI scans guided the aetiological diagnostic process in our cases of lissencephaly, CRASH syndrome, syntelencephaly, congenital cytomegalovirus infection, and leukodystrophy.
The diagnostic yield of genetic studies is increasing and these studies are becoming the first step in the diagnostic process. In children with GDD/ID, the CGH array is diagnostic in 7.8% of all cases, reaching 10.6% in cases with syndromic features. High resolution karyotyping result is abnormal in at least 4.6% of cases and in 18.6% of cases with syndromic features. Results from FMR1 for fragile-X screening are positive in at least 2% of patients with mild or moderate GDD/ID. MECP2 gene analysis provides a diagnosis in 1.5% of girls with moderate or severe GDD/ID, compared to less than 0.5% of boys with GDD/ID.8
CHG array is the genetic test with the highest diagnostic yield in children with GDD/ID with no established cause.8 It provided the diagnosis in 29.5% of the cases in our series (21 out of 71).
Next-generation (massive) sequencing panels will certainly change diagnostic approaches to determining the aetiologies of ID.15
Cost-effectiveness of complementary studies is apparently low in absence of clinical suspicion. The problem here is determining whether we perform too many studies, although identifying major problems, such as fragile-X syndrome, is necessary. Fifteen cases were identified among the 468 cases studied here. Early identification of vertically transmitted disorders, such as myotonic dystrophy or Duchenne muscular dystrophy, is very important from the diagnostic point of view.
Absence of anomalies (negative findings) should also be considered in the diagnostic process.5
This is a retrospective study of the diagnostic strategy we followed until only recently. We believe that it is important to analyse our practices and share our experience. While this population is heterogeneous and includes cases of easy diagnosis and regression, we did intend to avoid a selection bias.
GDD/ID with no established cause is observed in most genetically determined cases, and its social, family and economic impact is considerable. As a result, there is a high demand for early diagnosis. Most cases of GDD/ID are due to rare diseases and the European Union recommends that its member states implement national plans for rare diseases.16 The second strategic line of Spain's National Plan is early prevention and detection.17
After this review, we have decided to modify our evaluation strategy and emphasise the fact that in determining diagnoses, follow-up studies and personalised approaches are needed in addition to diagnostic protocols.
For children older than 2 to 3 years with isolated GDD/ID, with or without ASD and in the absence of progressive changes, we have decided to perform only one basic blood test (including muscle enzymes in boys to identify cases of Duchenne muscular dystrophy), homocysteine test, CDT, and thyroid hormone test. In children younger than 2 to 3 years in whom progressive changes can be expected, we will continue to run a comprehensive neurometabolic study with the aim of identifying IEM cases early on.
Genetic studies, including high-resolution karyotyping and fragile-X screening, will be performed on every GDD/ID case lacking a diagnosis. Exceptions would be fragile-X screening for girls with obvious macrocephaly or multiple malformations. We will perform CGH array in presence of certain factors such as family history of ID, miscarriages, intrauterine growth restriction, micro- or macrocephaly, microsomy or macrosomy, dysmorphic features, or any other associated malformations of the heart, kidneys, or eyes.
Brain MRI scans will be performed in cases of epilepsy, focal neurological signs, hypoacusia, visual impairment or ophthalmological alterations, macrocephaly/accelerated head growth and microcephaly, or decelerated head growth.
To conclude, we would like to highlight the importance of being able to diagnose rare diseases that are treatable. However, even when treatment is not possible, providing an aetiological diagnosis is always important for purposes of calculating risk of recurrence, providing genetic counselling, and assigning potential prenatal diagnoses. It can also be instrumental in resolving questions from families and professionals and also in limiting the number of diagnostic tests.
Conflicts of interestThe authors have no conflicts of interest to declare.
Please cite this article as: López-Pisón J, García-Jiménez MC, Monge-Galindo L, Lafuente-Hidalgo M, Pérez-Delgado R, García-Oguiza A, et al. Nuestra experiencia en el diagnóstico etiológico del retraso global del desarrollo y discapacidad intellectual: 2006-2010. Neurología. 2014;29:402–407.
