





La neuromielitis óptica es una enfermedad desmielinizante inflamatoria del sistema nervioso central, previamente considerada una variante de esclerosis múltiple, con la cual guarda semejanzas así como diferencias. Esta actualización tiene por objetivo efectuar una revisión histórica de la enfermedad, repasar sus características clínicas principales, brindar una actualización sobre los conocimientos de su fisiopatogenia, así como brindar una aproximación diagnóstica y terapéutica de esta entidad poco frecuente pero de una elevada morbimortalidad.
Neuromyelitis optica is a demyelinating inflammatory disease of the central nervous system, previously considered to be a variant of multiple sclerosis, from which it bears some resemblance as well as some differences. The objective of this update is to present an analysis of the epidemiology and history of the disease, its main clinical features, and to offer a review of the most recent knowledge concerning its pathogenesis, as well as bringing about a diagnostic and therapeutic approach to this disease, that despite being uncommon entails great morbidity and mortality.
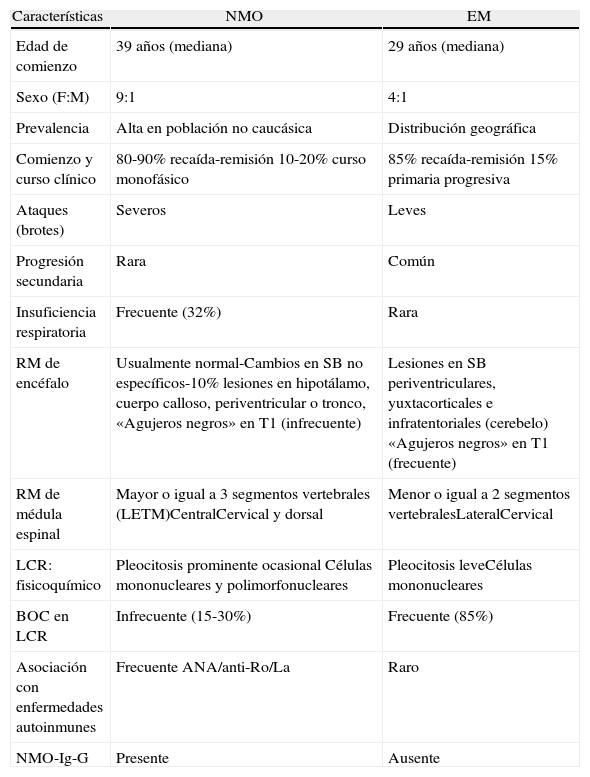
La neuromielitis óptica (NMO, o síndrome de Devic) es una enfermedad desmielinizante, autoinmune e inflamatoria crónica del sistema nervioso central (SNC) caracterizada por afectar severamente la médula espinal y a los nervios ópticos de forma monofásica o en brotes y remisiones, siendo una causa de discapacidad en jóvenes y adultos1−3. La primera descripción fue realizada por Sir Clifford Albutt en 1870, pero Eugene Devic y Fernand Gault fueron quienes en 1894 caracterizaron tanto la clínica como los procesos patológicos de esta entidad cuando publicaron los casos de 17 pacientes con asociación de neuritis óptica (NO) y mielitis transversa aguda (MTA) en forma simultánea o separadas por escaso tiempo3. Durante años esta afección ha sido clasificada como variante opticoespinal (asiática) de la esclerosis múltiple (OSMS)21. Sin embargo, actualmente existen variadas características clínicas, de laboratorio, neuroimágenes y en la anatomía patológica que la distinguen de la esclerosis múltiple (EM) (tabla 1)4,5,20. Principalmente la presencia de un autoanticuerpo específico en sangre llamado IgG-NMO o anti-AQP4 que se une a los canales de agua diseminados en el SNC llamados acuaporina-4 (AQP4): en los pies de los astrocitos y tiene un papel fundamental en la patogenia de esta enfermedad3,6.
Diferencias entre neuromielitis óptica y esclerosis múltiple
| Características | NMO | EM |
| Edad de comienzo | 39 años (mediana) | 29 años (mediana) |
| Sexo (F:M) | 9:1 | 4:1 |
| Prevalencia | Alta en población no caucásica | Distribución geográfica |
| Comienzo y curso clínico | 80-90% recaída-remisión 10-20% curso monofásico | 85% recaída-remisión 15% primaria progresiva |
| Ataques (brotes) | Severos | Leves |
| Progresión secundaria | Rara | Común |
| Insuficiencia respiratoria | Frecuente (32%) | Rara |
| RM de encéfalo | Usualmente normal-Cambios en SB no específicos-10% lesiones en hipotálamo, cuerpo calloso, periventricular o tronco, «Agujeros negros» en T1 (infrecuente) | Lesiones en SB periventriculares, yuxtacorticales e infratentoriales (cerebelo) «Agujeros negros» en T1 (frecuente) |
| RM de médula espinal | Mayor o igual a 3 segmentos vertebrales (LETM)CentralCervical y dorsal | Menor o igual a 2 segmentos vertebralesLateralCervical |
| LCR: fisicoquímico | Pleocitosis prominente ocasional Células mononucleares y polimorfonucleares | Pleocitosis leveCélulas mononucleares |
| BOC en LCR | Infrecuente (15-30%) | Frecuente (85%) |
| Asociación con enfermedades autoinmunes | Frecuente ANA/anti-Ro/La | Raro |
| NMO-Ig-G | Presente | Ausente |
BOC: bandas oligoclonales; EM: esclerosis múltiple; LCR: líquido cefalorraquídeo; LETM: mielitis transversa longitudinal extensa (del ingles); NMO: neuromielitis óptica; RM: resonancia magnética nuclear; SB: sustancia blanca.
De Wingerchuk et al.3.
En esta revisión describiremos la epidemiología, la inmunopatogenia, las características clínicas, los criterios diagnósticos, los métodos complementarios y el tratamiento de esta enfermedad de acuerdo a los múltiples avances e investigaciones que aumentaron el interés de los neurólogos en las últimas décadas.
Epidemiología y aspectos genéticosLa NMO ha sido publicada en todos los continentes y razas. La prevalencia estimada es de 0,3-4,4 por 100.000 habitantes14. Asimismo, es una causa de enfermedad desmielinizante relativamente común en poblaciones no blancas: afrobrasileños (15%), en individuos japoneses (20-30%), oeste de India (27%), asiáticos del este como China (36%), Singapur (48%) e India (10-23%)3,15,34 del total de enfermedades desmielinizantes del SNC16,17 con menor prevalencia en pacientes caucásicos (Estados Unidos, Canadá y Europa) llegando a un 1,5-2%14. Cabe aclarar que en Asia y Latinoamérica su prevalencia sería mayor6,18. Datos argentinos señalan un 7,5% de enfermedad desmielinizante3,14,18. La edad media de inicio es de 39 años, puede presentarse en población pediátrica y en ancianos, y es 9 veces más frecuente en mujeres que en hombres19,50,51. Durante el último trimestre del embarazo y en el posparto existe un riesgo aumentado de recaída14. No se han comunicado casos de transmisión al feto14. Alrededor de un 3% de los pacientes con NMO tiene familiares con esta enfermedad14,52. Aunque no se conoce la susceptibilidad genética, está claro que existe una prevalencia aumentada en la población asiática; debido a esto se realizó un estudio en Japón que analizó las características genéticas de la OSMS que fue comparable con la NMO y concluyó que la esclerosis múltiple (EM) típica estuvo asociada a HLA-DRB1*1501, mientras que la OSMS/NMO se asoció a HLA-DPB1*050111,22,35. Sin embargo, el 60% de la población japonesa expresa el alelo HLA-DPB1*050121. Por ello, otro estudio comparó pacientes con OSMS/NMO con IgG-NMO positivo y negativo, y se observó que aquellos pacientes con anticuerpo positivo presentaban manifestaciones severas de la enfermedad, así como también elevada tasa de dicho alelo en comparación con IgG-NMO negativo23. Esta asociación fue encontrada también en pacientes chinos. Sin embargo, el alelo HLA-DRB1*03 fue encontrado en pacientes brasileños mulatos con NMO36 y afrocaribeños37. La OSMS parece ser una entidad similar a la NMO occidental. Sin embargo, no está claro si las diferencias de OSMS y NMO entre Asia y los países occidentales se deben a diferencias biológicas o de nomenclatura54. Finalmente, es conocida la asociación con enfermedades autoinmunes organoespecíficas como la miastenia gravis y la tiroiditis autoinmune, y no organoespecíficas como el lupus eritematoso sistémico (LES) y síndrome de Sjögren (SS)24. El anticuerpo que se encontró con mayor frecuencia en un estudio que evaluó NMO y mielitis con afectación de 3 o más segmentos medulares (mielitis transversa longitudinal extensa [LETM]) fue el antinuclear (43,8%) y el anti-Ro/anti-La (15,7%). En este estudio se encontró una coexistencia de NMO y enfermedad autoinmune en el 28% de los pacientes24.
InmunopatogeniaEn el año 2004 se descubre la presencia de una inmunoglobulina G específica, cuyo blanco es la AQP4, dando lugar a la noción de que la NMO constituía en realidad una entidad completamente distinta a la EM2,6.
Las acuaporinas (AQP) son proteínas transmembrana con amplia representación en todo el organismo. Se conocen 13 tipos de AQP, las cuales se dividen en AQP ortodoxas (solo permeables al agua) y acuagliceroporinas (permeables al agua, a la urea y al glicerol)38. La AQP4 se halla en máximas concentraciones en el SNC (donde se ha determinado su presencia en la corteza cerebral y cerebelosa, en porción posterior del nervio óptico y en las células de Müller de la retina; en el epéndimo, hipocampo y médula espinal), y en concentraciones similares en la médula renal. Asimismo, diversos estudios demostraron su presencia en otros tejidos de la economía39.
En el SNC, la AQP4 se expresa en los astrocitos (principalmente en los procesos astrocíticos en contacto con los vasos sanguíneos), y se halla particularmente concentrada en las superficies piales y ependimarias en contacto con el líquido cefalorraquídeo (LCR). La AQP4 es una proteína cuyos monómeros se componen de 6 segmentos que abarcan la membrana y 2 segmentos helicoidales. Se presenta en 2 isoformas principales, M1 y M23, las cuales se asocian en las membranas formando heterotetrámeros. A nivel de la membrana plasmática los tetrámeros de AQP4 adoptan una disposición en diseño ortogonal (OA, del inglés ortogonal arrays); se ha observado in vitro que las isoformas M23 participan de grandes OA, mientras que la isoforma M1 forma tetrámeros que se hallan poco concentrados y no tienen la capacidad de formar OA si no coexisten con la presencia de AQP4-M23. Asimismo, la proporción M1/M23 en un OA determina el tamaño del mismo (mayor tamaño a mayor proporción de M23). La AQP4 interviene en la regulación del flujo transmembrana de agua. Otras funciones fueron estudiadas en ratones genéticamente desarrollados con ausencia del gen AQP4 (AQP4−) y se observó que eran menos proclives a desarrollar edema citotóxico, pero que a su vez tenían mayor retención hídrica ante edema vasogénico; se concluyó entonces que, si bien no afectaba a la morfología y a la proliferación astrocitaria, en ratones AQP4− la migración astrocitaria y la gliosis cicatricial se hallaba afectada, por lo tanto, los astrocitos AQP4− demostraron menor capacidad de absorción de potasio desde el líquido extracelular, lo cual se manifestó como alteración en los sentidos de estos roedores (demostrado por potenciales evocados auditivos con mayor umbral de respuesta y potenciales pequeños en el electrorretinograma y electroolfatograma)40.
Como prerrequisito común a todas las enfermedades humorales autoinmunes, la AQP4 debe ser presentada a un linfocito T Helper (LtH). Se han descripto distintos epítopes antigénicos en la estructura de la AQP4, ante los cuales los LtH reaccionarían, dando lugar a la diferenciación de linfocitos B a plasmocitos productores de anticuerpos anti-AQP441,42. La apoptosis y fagocitosis de los restos celulares por parte de macrófagos o microglía puede ser el primer paso, ya que estas podrían presentar epítopes inmunogénicos a linfocitos. Un estudio describió una mayor proporción de LtH 17 (TH17), un subtipo de linfocitos CD4+, en pacientes con NMO, en línea con las elevadas concentraciones de IL17. En este estudio se observó también la presencia de linfocitos CD4+ que demostraron reacción cruzada ante un transportador de membrana del Crostridium perfringens, abriendo la posibilidad de que pudiera tratarse de una enfermedad de reacción cruzada43. Por lo tanto, existen 2 hipótesis respecto al rol de los LtH: podría tratarse de la persistencia de LtH autorreactivos producto de una deficiente selección negativa de linfocitos autorreactivos, o de un fenómeno de reacción cruzada ante un xenoantígeno.
Los anticuerpos anti-AQP4 son sintetizados principalmente fuera del SNC, con una concentración en plasma 500 veces superior a la del LCR. Los anticuerpos anti-AQP4 se unen con alta afinidad al tercer dominio extracelular de la AQP4. Sin embargo, la afinidad es mucho mayor hacia los OA y es por ello que la unión de anticuerpos anti-AQP4 es mayor cuanto mayor es la proporción del isotipo AQP4-M23 sobre AQP4-M1. Al producirse la unión a su antígeno, se dan los siguientes efectos: a) disfunción de la AQP4; b) internalización de la AQP4; c) activación del complemento, y d) activación de células efectoras (principalmente linfocitos natural killers)44. El 98% de estos anticuerpos son del subtipo IgG1, con notable capacidad para activar el complemento; es por esto que los anticuerpos anti-AQP4 ejercen su efecto astrocitotóxico primordialmente mediante la activación de complemento. Una vez activadas las citocinas (IL17, interleucina B, factor estimulante de colonias de granulocitos) se reclutan neutrófilos y eosinófilos45, cuya degranulación induce muerte astrocitaria. Esto provoca lesión de los oligodendrocitos con el consiguiente daño axonal y, por fenómenos de degeneración retrógrada, la muerte neuronal. El paso final de la cascada es la infiltración de macrófagos. Toda esta secuencia inflamatoria aumenta la permeabilidad de la barrera hematoencefálica (BHE), facilitando el paso de anticuerpos anti-AQP4 y profundizando el proceso inflamatorio. Los anticuerpos anti-AQP4 adicionalmente pueden causar citotoxicidad en presencia de linfocitos natural killer (LNK), ante la ausencia de complemento14. Un estudio reciente describe que el grado de activación de complemento podría depender notablemente de la disposición en OA, independientemente de la afinidad de los anticuerpos anti-AQP4 a estos, siendo significativamente menor la activación del complemento cuando los anticuerpos anti-AQP4 no están unidos a OA. Esto podría deberse a una interacción multivalente del factor C1q con los anticuerpos anti-AQP4 cuando estos se hallan unidos a OA. Por otro lado, la citotoxicidad mediada por LNK no tendría relación con la proporción de anticuerpos anti-AQP4 unidos a OA46.
Además de la disposición de la AQP4, la permeabilidad relativa de la BHE en ciertas regiones condiciona una mayor vulnerabilidad a dichos anticuerpos (anti-AQP4), como es el caso de la región prelaminar de la cabeza de los nervios ópticos. A nivel de la médula espinal, se demostró una mayor concentración de AQP en los astrocitos fibrosos de las 2 láminas más superficiales del asta posterior medular. Finalmente, la distribución ependimaria sumada a una mayor permeabilidad en dicha región da lugar a la aparición de lesiones periventriculares y periependimarias medulares47. La permeabilidad de la BHE se incrementa en procesos inflamatorios sistémicos y esto tiene correlato con la frecuente asociación entre procesos infecciosos (principalmente virales), inmunizaciones y brotes de NMO. Según un estudio reciente, se registraron antecedentes de procesos infecciosos en 18% seronegativos y 29% seropositivos. En este mismo estudio, en el que se incluyeron 175 pacientes (137 seropositivos y 38 seronegativos) se mostró que la seronegatividad para anti-AQP4 tendría un correlato clínico: los pacientes seropositivos eran predominantemente de sexo femenino, tenían coexistencia de cuadros inflamatorios sistémicos, presentaban ataques más severos, mayor disminución de agudeza visual durante episodios de NO, mayor frecuencia de síntomas motores y lesiones más extensas a nivel medular. En los pacientes seronegativos, la NO bilateral con o sin mielitis concomitante se observaba más frecuentemente, así como un curso monofásico48. Esto apoyaría la hipótesis de un autoantígeno diferente al de los pacientes seropositivos: en pacientes seronegativos para IgG anti-AQP4, descartando la sensibilidad y especificidad de los diversos métodos de determinación, la AQP4 puede no ser el blanco inmunogénico.
A nivel histopatológico es característica la necrosis y la cavitación de las lesiones, así como la hialinización de pequeños vasos y los infiltrados perivasculares de eosinófilos, neutrófilos y, en mucho menor número, linfocitos T CD4+. Empleando métodos de inmunohistoquímica, es posible visualizar el depósito perivascular de IgG anti-AQP4 y complemento49.
Características clínicasEsta enfermedad se presenta clínicamente con NO y MTA en forma polifásica (de recaída) en el 60% de los casos al año y en el 90% a los 3 años, siendo menos frecuente el curso monofásico (simultáneo o no, asociación de NO y MTA con un lapso < 30 días)7−9,11. Estos hallazgos, junto con exámenes complementarios por RM y el anticuerpo anti-AQP4, son la base de los criterios diagnósticos para NMO (tabla 2)4,5,7,25,26 Generalmente la NO precede a la MTA en meses o años7,11. Sin embargo, cuando el curso es monofásico típicamente están separados solo por escasos días y es más frecuente en jóvenes7,11,31. La NO se caracteriza por pérdida de la agudeza visual, dolor a la movilización ocular y discromatopsia en uno o ambos ojos. Generalmente es unilateral y luego rápidamente se afecta el contralateral8,9. El campo visual revela escotoma central y el fondo ojo puede ser normal o patológico (edema, atrofia o papila pálida). La ceguera ocurre en el 60% de las formas recurrentes y en el 22% de las monofásicas7−9. En tanto la MTA se presenta con para o tetraplejía, nivel sensitivo y alteración de esfínteres7,9. El dolor radicular, los espasmos tónicos paroxísticos (recurrentes, dolorosos y con una duración entre 20-45 s) y el signo de Lhermitte se dan en el 33% de las formas recurrentes7,9. Los segmentos cervicales y dorsales altos en forma de LETM son los más frecuentemente afectados13. El hipo y las náuseas persistentes e intratables se pueden presentar en el 17-43% y la afectación respiratoria puede generar paro y muerte por extensión al tronco del encéfalo en un tercio de los pacientes4,10,11. La consecuencias clínicas de las recaídas en la NMO en más del 50% se deben a una agudeza visual < 20/200 y a paraplejía que generan secuelas severas4,32,33.
Comparación de los criterios diagnósticos propuestos para la neuromielitis óptica
| Wingerchuk et al., 1999 | Wingerchuk et al., 2006 | Criterios NMSS, 2008 |
| NMO definitiva | NMO definitiva | NMO definitiva |
| Criterios absolutos: | Criterios absolutos: | Criterios absolutos |
| 1. NO | 1. NO | 1. NO |
| 2. MTA | 2. MTA | 2. MTA |
| 3. Sin enfermedad fuera del | Al menos 2 criterios de apoyo: | Hiperintensidad en T2 > 3 |
| nervio óptico y la médula espinal | 1. RM de cerebro inicial que no | segmentos vertebrales e |
| Al menos 1 criterio mayor | cumpla criterios para EM | hipointensidad en T1 durante la |
| 1.RM de cerebro que no | 2. RM medular con una lesión | MTA |
| cumpla criterios para EM | de 3 o más segmentos | Sarcoidosis, vasculitis o LES |
| 2. RMmedular con una lesión | vertebrales | (clínicamente manifiesta) |
| en T2 de 3 o más segmentos | 3. Seropositividad para NMO-IgG | excluye el diagnóstico de NMO |
| vertebrales | Spectrum de NMO (2007) | Al menos un criterio de apoyo: |
| 3. Pleocitosis > 50 cél./mm3 o > 5 | 1. Episodios idiopáticos simples o | 1. RM de cerebro inicial que no |
| neutrófilos/mm3 en LCR | recurrentes de LETM en RM | cumpla criterios para EM |
| Al menos 2 criterios menores | 2. NO recurrente o simultánea | 2. Seropositividad para NMO-IgG |
| 1. NO bilateral | bilateral | |
| 2. NO severa con agudeza visual | 3. OSMS en asiáticos | No están incluidos los |
| fija < 20/200 en un ojo | 4. NO o LETM asociadas a | criterios para Spectrum de |
| 3.Debilidad severa y fija | enfermedades autoinmunes | NMO |
| relacionada a un ataque en 1 o | 5. NO o LETM asociada con | |
| mas miembros | lesiones típicas de NMO | |
| (hipotálamo, cuerpo calloso, | ||
| periventricular o tronco) |
EM: esclerosis múltiple; LES: lupus eritematoso sistémico; LETM: mielitis transversa longitudinal extensa (del inglés); MTA: mielitis transversa aguda; NMO: neuromielitis óptica; NMSS: National Multiple Sclerosis Society; NO: neuritis óptica; OSMS: esclerosis múltiple opticoespinal; RM: resonancia magnética.
El descubrimiento de los anticuerpos IgG-NMO ayudó a definir el espectro clínico (formas limitadas de NMO) relacionado a esta enfermedad (tabla 2)6. Asimismo, dichos anticuerpos se detectan en un alto porcentaje de los pacientes con un primer evento (inicio de la enfermedad) de la NMO. Es por esto que ante un primer ataque de LETM y/o NO con IgG-NMO positiva existe un riesgo elevado de recurrencia, vale decir, de conversión a NMO definida27,32,33. Estos conceptos fueron estudiados por el grupo de la Clínica Mayo en un estudio prospectivo de pacientes con un primer evento (síndrome clínico aislado) de LETM e IgG-NMO positivo, encontrándose un 56% de riesgo de recurrencia de LETM o NO (conversión a NMO) durante el seguimiento a un año27. El 20% de los pacientes con NO recurrente son IgG-NMO seropositivos y dichos pacientes tienen un 50% de riesgo a desarrollar mielitis transversa y resultados desfavorables a nivel visual28. En un estudio de cohorte japonés se encontró que el 58% de los pacientes con OSMS fueron seropositivos en comparación con 0% de las EM típicas6,29. Por lo tanto, la positividad para IgG-NMO es de aproximadamente un 50% en pacientes con recidiva de LETM y de un 25% en pacientes con NO simultánea o recurrente con RM de encéfalo normal (excluyendo los nervios ópticos)6,27,28,30. Dentro del espectro de la NMO se encuentra la asociación con enfermedades autoinmunes no órgano-específicas y órgano-específicas24. Estudios recientes demostraron que los pacientes con LES o SS clínicamente definido carecen de seropositividad para IgG-NMO así como también, de síntomas clínicos del espectro NMO30,24. Por lo tanto, la asociación de NMO seropositiva con enfermedades autoinmunes (LES o SS) representa una coexistencia de 2 enfermedades autoinmunes más que una complicación vasculítica secundaria a la enfermedad sistémica autoinmune3,6,14,30,24.
Estudios complementariosTanto la utilización de métodos complementarios como la de un algoritmo diagnóstico es fundamental no solo para poder certificar la sospecha de NMO, sino también para poder descartar otras enfermedades que presentarán un tratamiento específico.
Resonancia magnéticaEl advenimiento de la RM y los avances en la calidad de las imágenes durante las últimas 2 décadas produjo un mayor interés en el diagnóstico de NMO y se convirtió en un método fundamental, formando actualmente parte de los actuales criterios diagnósticos. La RM de médula espinal realizada en los primeros días a semanas posterior al ataque agudo de mielitis se caracteriza por presentar edema y captación de contraste21. El hallazgo de LETM es considerado parte fundamental de los criterios diagnósticos para el diagnóstico de NMO junto a la detección de los anticuerpos IgG-NMO. Un análisis de los componentes individuales de dichos criterios reveló que la RM que muestra lesiones de 3 o más segmentos vertebrales tiene una especificidad de 83% y una sensibilidad del 98% para el diagnóstico de NMO54,102. Si bien estas lesiones extensas son características, pueden aparecer lesiones pequeñas, las cuales son menos específicas55. Lennon et al. reportaron que el 60% de los pacientes con LETM recurrente eran seropositivos para NMO-IgG6. Además, en los pacientes con mielitis transversa incompleta y lesiones pequeñas (menos de 3 segmentos) la detección de IgG-NMO es infrecuente56. Hay que tener en cuenta que estas lesiones extensas pueden evolucionar a cavitaciones centrales y atrofia de la médula espinal57.
En el pasado, se ha discutido mucho sobre la presencia de lesiones en RM de encéfalo fuera de los nervios ópticos. Sin embargo, estudios recientes han reportado una alta frecuencia de alteraciones cerebrales en la RM de estos pacientes. Durante el seguimiento se observan lesiones en la RM cerebral en el 60-88% de los pacientes12,58. Las lesiones asintomáticas son más comunes, aunque pueden existir lesiones sintomáticas en la evolución de la enfermedad. Pittock et al., en una revisión de 60 pacientes (68% seropositivos para IgG-NMO), informaron que el 60% tenía lesiones cerebrales. La mayoría eran inespecíficas y no fueron consideradas típicas de EM. Seis pacientes (10%) tenían lesiones «EM-like» y 4 cumplían con los criterios Barkoff. Cinco (8%) presentaron lesiones de localización atípica para EM (tálamo, hipotálamo y lesiones extensas de sustancia blanca)12. Otros reportes han demostrado también la aparición de lesiones hipotalámicas, algunas con endocrinopatías asociada59. Una revisión sistemática de imágenes en RM de encéfalo en 120 pacientes IgG-NMO positivos mostró que el 6% de los sujetos tenían lesiones adyacentes a los ventrículos (principalmente tercero y cuarto ventrículos). La distribución de estas lesiones cerebrales concuerda con la localización de los canales de agua AQP412.
Es interesante comentar que en una serie de casos98 (19 autopsias analizadas por inmunohistoquímica) de NMO y su espectro en una cohorte transversal se visualizó ausencia de desmielinización cortical cerebral y cerebelosa con conservación de la distribución de la AQP4. En otro estudio99 que evaluó la presencia de lesiones corticales (in vivo) en secuencias de inversión-recuperación y espesor cortical por la aplicación Freesurfer en T1 (volumetría por eco) sobre 90 pacientes (NMO: 30; EMRR: 30; controles normales: 30) no se encontraron lesiones corticales en los controles normales ni en los pacientes con NMO. Sin embargo, 20/30 (66,7%) pacientes con EM sí las presentaban. Dichos hallazgos fueron corroborados en otro estudio100 que comparó NMODS (n=10) vs EM (n=18) en RM de 7 Tesla y se encontró que las lesiones en sustancia blanca de la EM eran perivenulares (92%) en comparación a las de NMOSD (35%) que eran «paravenulares» con ausencia de trastorno cortical en la sustancia gris (NMOSD: 0 vs EM: 7/18). Por lo tanto, el análisis de la corteza por RM es una herramienta útil fundamentalmente para el diagnóstico diferencial con la EM.
Líquido cefalorraquídeoEl estudio del LCR durante un episodio agudo puede mostrar pleocitosis con predominio de polimorfonucleares (neutrófilos y eosinófilos) de 50 a 1.000 células por mm3, lo que diferencia de la EM donde la pleocitosis raramente excede 50 células por mm3. La detección de BOC es infrecuente (< 30%).7,53 Los niveles bajos de IgG1 en LCR en comparación con EM indica menor respuesta T helper de tipo 1 (Th1)60. No se encontraron diferencias en los niveles de citocinas y quimiocinas en NMO en comparación con los pacientes de EM61.
Otros métodos complementariosActualmente se reconoce una fuerte asociación de la NMO con enfermedades autoinmunitarias como fue comentado anteriormente con una alta asociación del anticuerpo antinuclear62.
El estudio de los potenciales evocados visuales (PEV) es ampliamente utilizado para demostrar afectación del nervio óptico. Además, la identificación de los patrones del PEV en pacientes con NMO podría ayudar a diferenciar esta enfermedad de la EM. Silvio et al. demostraron mediante un estudio en 19 pacientes (38 ojos examinados) con diagnóstico definitivo de NMO un patrón que es diferente al encontrado en la EM. El 81,6% de los ojos examinados se caracterizó por presentar ausencia de respuesta o disminución de la amplitud de la onda P100 con latencia normal101.
Mediante la tomografía de coherencia óptica (TOC) se puede medir espesor de la capa de fibras nerviosas de la retina (CFNR)63. Existe una correlación en estudios histológicos entre el recuento axonal del nervio óptico y el espesor de la CFNR medida por TOC. También pueden observarse cambios significativos en el espesor de la retina en pacientes con EM sin episodios visuales comparados con controles sanos. Esto ha llevado a proponer este método como un posible biomarcador de degeneración axonal en EM64.
Naismith et al. reportaron una disminución significativa del espesor de la CFNR en ojos no afectados clínicamente de pacientes con NMO. Demostraron además que después de un episodio remoto de NO se evidencia CFNR más delgada en la NMO comparada con la EM después de ajustar las muestras (agudeza visual y número de recaídas). Este hallazgo es compatible con una mayor pérdida axonal en los sujetos con NMO. A pesar de estos datos todavía no está del todo clara la utilidad de la TOC como marcador pronóstico y de respuesta terapéutica65.
TratamientoEl tratamiento de la NMO comprende 2 instancias: el tratamiento agudo de los brotes y el tratamiento inmunosupresor destinado a reducir la frecuencia de brotes y la progresión de la discapacidad. No se dispone de ensayos controlados aleatorizados que evalúen el tratamiento de los brotes en la NMO. Como tratamiento agudo, la intervención inicial más difundida es la administración de metilprednisolona intravenosa (MEV), 1 g/día por 3 a 5 días. Esta indicación surge de experiencia empírica y de la evidencia de la respuesta observada en mielitis asociadas a EM. En aquellos casos en los que no se observe mejoría clínica significativa con esta intervención, la plasmaféresis (PF) puede ser ofrecida; la evidencia para esta conducta surge de un estudio en el que 2 pacientes con mielitis asociada a NMO recibieron PF por no demostrar mejoría con un curso estándar de MEV, y tras la PF demostraron mejoría significativa66. Posteriormente numerosas series de casos (una de ellas que incluyó 23 pacientes que tras recibir MEV recibieron PF por falta de respuesta67) brindaron evidencia de que la PF fue efectiva en pacientes con MTA (espectro de la NMO) que no tuvieron respuesta al tratamiento con MEV. Una revisión reciente de la técnica de la PF y la bibliografía (experiencia en el uso de PF en brotes de NMO) resume la evidencia para ataques medulares, ópticos y cerebrales; afirmando que el empleo de PF en ataques medulares se asocia con menor EDSS (Expanded Disability Severity Score) residual. Asimismo, la reducción del EDSS (EDSS durante brote-EDSS luego del tratamiento) era mayor en pacientes tratados con PF en su primer evento clínico. La evidencia es conflictiva respecto al beneficio en relación con el inicio temprano: un estudio sugirió beneficio con tratamiento temprano mientras que los resultados de otro, no. La respuesta no varía de acuerdo al estado serológico (seropositivo o seronegativo) según los estudios mencionados y el único predictor válido de beneficio fue la presencia de reflejos normales o reflejos vivos durante el brote. La evidencia que indica beneficio asociado al uso de PF impide el diseño de un estudio controlado por placebo, porque este no cumpliría con los criterios metodológicos (por ejemplo, la aprobación por el comité de ética). Respecto a los ataques de NO, reuniendo la casuística de NO severas (39 NO con agudeza visual menor a 1/10) se observó que el tratamiento precoz se asociaba con mejoría significativa y que el beneficio reportado en aquellos pacientes tratados con PF en forma tardía (lo cual se asoció con menor beneficio en comparación a quienes recibieron PF precozmente) se beneficiaron en grado similar a los paciente tratados solo con MEV. Finalmente, respecto a los episodios desmielinizantes cerebrales asociados a NMO, la evidencia es escasa pero indica un efecto beneficioso68. Cabe mencionar otro estudio que concluyó que una menor puntuación EDSS previa al brote y la ausencia de atrofia medular fueron predictores de mayor beneficio asociado al tratamiento con PF69. Un estudio alerta acerca de que los niveles de anticuerpos anti-AQP4, tras sufrir una reducción drástica durante la PF, pueden presentar un rebote y elevarse rápidamente a niveles incluso superiores a los previos, por tanto recomiendan iniciar la inmunosupresión concomitante con la administración de PF70.
La eficacia de la gammaglobulina intravenosa (GIV) en la NMO no ha sido demostrada. Un estudio controlado por placebo diseñado para evaluar la eficacia de la GIV en pacientes con déficit visual reciente debido a NO asociada a NMO no demostró beneficio71. Adicionalmente, no hay evidencia acerca de la eficacia en brotes medulares. Aun así, un artículo muy reciente de Dean Wingerchuk revisa la fisiopatogenia de la enfermedad y poniendo en consideración que la NMO es un trastorno mediado por inmunidad humoral, propone la GIV como tratamiento en aquellos pacientes donde sería arriesgado administrar MEV (por ejemplo, diabéticos mal controlados) o PF (inestabilidad hemodinámica, falta de accesos vasculares periféricos)72.
Respecto al tratamiento orientado a reducir la frecuencia de brotes, una revisión reciente se extiende acerca de 6 terapias sobre las cuales la evidencia indica efectividad; estos fármacos son azatioprina, rituximab, mofetil micofenolato, metotrexato, mitoxantrone y corticoides orales73. Se revisarán las mismas y la bibliografía publicada tras esta revisión, así como tratamientos de dudosa efectividad o en desarrollo.
Excede el objetivo de esta revisión el repasar los mecanismos y efectos adversos de los corticoides como tratamiento prolongado, por tanto solo se discutirá brevemente su empleo como tratamiento preventivo en NMO. Utilizados para este fin aproximadamente desde 1970, la evidencia se sustentó inicial y principalmente en la amplia experiencia en su empleo. Un estudio presentado en 1999 observó la eficacia del tratamiento conjunto con azatioprina y prednisona74, pero como se verá más adelante, la eficacia de la azatioprina puede ser parcialmente responsable de esta eficacia. Un estudio retrospectivo75 muy interesante observó que en 9 pacientes con NMO que recibieron corticoides y luego los interrumpieron, la tasa de recaídas durante el tratamiento fue cercana al 33% en comparación con la tasa de recaídas fuera de tratamiento, lo cual parece indicar un efecto positivo modesto. Si bien el tratamiento con corticoides orales como primer y único fármaco es una práctica extendida, la experiencia es mucho más favorable con otros fármacos y los efectos adversos de los corticoides desaconsejan esta iniciativa. Por otro lado, su empleo como droga adyuvante se halla muy difundida y aceptada.
La azatioprina es una prodroga, cuya forma activa (6-mercaptopurina) es un análogo nucleósido que impide la síntesis de purinas, comprometiendo la maduración de linfocitos T y B. La primera evidencia acerca de la eficacia de la azatioprina surge en 1998 con un estudio que asoció azatioprina más corticoides en 7 pacientes con NMO, observando reducción en la puntuación EDSS y ausencia de brotes a lo largo de 18 meses de seguimiento, sin efectos adversos significativos74. Numerosas series de pacientes tratados con azatioprina reportaron similares resultados: dentro de ellas destaca un estudio prospectivo de 86 pacientes con NMO y 13 pacientes con episodios del espectro NMO con seropositividad. En esta serie solo 70 pacientes tuvieron más de 12 meses de seguimiento, de los cuales el 31% presentó estabilidad o mejoría del EDSS a pesar de presentar recaídas y 37% de ellos no presentaron recaídas por una mediana de 24 meses de seguimiento. Por otro lado, en 38 pacientes se debió suspender la azatioprina por falta de eficacia (13 pacientes), diversos efectos adversos (22 pacientes) o desarrollo de linfoma (3 pacientes, 1 de tipo Hodgkin y 2 no hodgkinianos)76. Además de las reacciones adversas más conocidas (diarrea, náuseas, pérdida de peso, hepatotoxicidad, leucopenia y supresión medular) se indica efectuar determinación de la actividad de la tiopurina metiltransferasa, ya que niveles de actividad bajos se asocian con un aumento considerable del riesgo de toxicidad y deben plantear el empleo de un fármaco alternativo.
El rituximab es un anticuerpo monoclonal anti-CD20, con múltiples indicaciones para diversas enfermedades autoinmunes y linfoproliferativas cuyo elemento en común es el rol preponderante del linfocito B activado. Numerosas series de casos indican consistentemente una reducción en la tasa de recaídas anuales y una reducción en la puntuación del EDSS (considerando que esta reducción puede deberse a la mejoría obtenida por el tratamiento agudo de distintos episodios desmielinizantes agudos que sufrieron los pacientes) y esta experiencia fue replicada en una serie de 8 pacientes pediátricos con NMO. Un estudio retrospectivo multicéntrico describió 2 regímenes empleados cotidianamente, uno empleado habitualmente para tratar linfoma (375mg/m2 semanalmente por 4 semanas) y otro empleado para el tratamiento de enfermedades autoinmunes (1.000mg inicialmente y 1.000mg a los 14 días). Se demostró reducción de la tasa de brotes anuales y reducción o estabilización del EDSS en 80% de los pacientes77. Tras numerosas series con resultados similarmente favorables78 surge un estudio en el año 2011 que describe su experiencia positiva con rituximab: la diferencia es que en este estudio se empleó como marcador de retratamiento el recuento de linfocitos CD27+ (a diferencia de estudios previos donde se empleó el recuento de linfocitos CD19+); este marcador permitió reducir el número de infusiones, abaratando costes sanitarios, sin relegar la calidad y eficacia79. Por otro lado, cabe mencionar un estudio retrospectivo publicado en el año 2012 donde 8 pacientes con NMO (la mitad seropositivos) y un paciente con LETM recurrente (seropositiva) fueron tratados con rituximab: 3 de ellos no presentaron recaídas en un seguimiento promedio de 22 meses, mientras que 6 de ellos continuaron con recaídas. Como crítica los autores describen que los pacientes que siguieron con recaídas llevaban menos de 6 meses de evolución y el 30% de ellos no había tenido tratamiento inmunosupresor previo (solo corticoides orales) pero es poca la literatura que reporta resultados negativos para rituximab en NMO80.
El micofenolato mofetil es un profármaco cuyo metabolito activo es el ácido micofenólico que impide la síntesis de novo de nucléotidos de guanosina, comprometiendo la síntesis de ADN y ARN primordialmente en linfocitos B y T. Fue en 2006 cuando se reportó el caso de una niña de 9 años que se mantuvo libre de brotes a lo largo de 2 años bajo tratamiento con micofenolato. Luego, en el año 2009, se presentó una serie de 24 casos (15 casos de NMO y el resto de trastornos del espectro NMO seropositivos), de los cuales 19 pacientes permanecieron bajo tratamiento por una mediana de 27 meses presentando una reducción de la tasa anual de recaídas, pero sin reducción significativa del EDSS promedio81.
El metotrexato es un inhibidor de la dihidrofolatorreductasa, una enzima crucial para la síntesis de bases purínicas y timidínicas, empleada en distintas enfermedades autoinmunes y oncológicas. Más allá de descripciones de casos, un estudio publicado en el año 2000 sobre 8 pacientes tratados con metotrexato y prednisona sugirieron su eficacia: en este artículo, 4 pacientes fueron tratados con metotrexato y prednisona, mientras que otros 4 fueron tratados con prednisona y ciclofosfamida. Todos los pacientes estabilizaron su puntuación EDSS tras iniciar tratamiento con prednisona y metotrexato82.
La experiencia es escasa acerca de la eficacia de mitoxantrone en NMO. Si bien es conocido su efecto beneficioso en EM de curso agresivo, la experiencia no se ha replicado en NMO. Además de un pequeño estudio de 3 pacientes con NMO tratados con mitoxantrone reportado en un trabajo de investigación acerca de la AQP que no ofrece cita y no pudo ser encontrado (de 3 pacientes con NMO, 2 presentaron reducción en la tasa de recaídas anual y uno aumentó la tasa de recaídas)83, 2 estudios señalan un efecto beneficioso del mitoxantrone en la NMO. El primero consiste en un estudio retrospectivo y prospectivo de 20 pacientes con NMO o trastornos del espectro NMO que tuvieron al menos 2 ataques en el año previo a iniciar tratamiento con mitoxantrone y que presentaron una reducción del 75% (mediana) de la tasa de recaídas anuales y un 50% de pacientes se mantuvieron libres de recaídas (seguimiento promedio de 41 meses)84. El segundo es un pequeño estudio prospectivo en el que 5 pacientes con NMO fueron tratados con mitoxantrone y a lo largo de 2 años de tratamiento 4 pacientes presentaron mejoría clínica y radiológica, a pesar de que 2 pacientes presentaron una recaída cada uno; el paciente que no demostró mejoría es porque falleció a los 4 meses de iniciado el estudio por una embolia pulmonar85.
Respecto a tratamientos potencialmente beneficiosos, pero con escasa evidencia, un ensayo no controlado de GEV que evaluó la reducción de episodios desmielinizantes en pacientes con trastornos del espectro NMO (5 con NO recidivante y/o mielitis y 3 con LETM recurrente) demostró reducción de la tasa de recaídas respecto a los años previos en cada paciente con reducción del EDSS en 5 pacientes86. En otro artículo se reporta el caso de un paciente con NMO seropositivo que obtuvo respuesta favorable (reducción de dolor neuropático, del EDSS y de los valores de anti-AQP4) al recibir tocilizumab (un anticuerpo anti-IL6)87. Por otro lado, un estudio informa que el empleo de un anticuerpo anti-AQP4 recombinante (el cual no tiene capacidad de activación del complemento ni induce citotoxicidad mediada por células) que compite con el anticuerpo anti-AQP4 presente en pacientes con NMO, impidió el desarrollo de lesiones en un modelo in vitro de médula espinal y en un ratón, abriendo la puerta a una nueva generación de tratamientos con prometedor futuro88. Finalmente, solo 2 casos reportados en sendos estudios indican eficacia de glatiramer acetato89.
La evidencia señala que el natalizumab90 y el interferón beta91–93 no son efectivos. Además, este último puede aumentar la tasa de brotes. Se ha reportado un paciente con un trastorno del espectro NMO que recibió tratamiento con fingolimod durante el estudio TRANSFORMS y que desarrolló extensas lesiones bihemisféricas, frontales y parietales con edema vasogénico y realce con contraste94.
Finalmente cabe mencionar que se encuentra en completo un estudio no ciego diseñado para evaluar eficacia y seguridad del eculizumab (autoanticuerpo antiproteína C5 del complemento), del cual no se han publicado resultados aún95. Asimismo, se halla próximo a iniciar la selección un estudio fase i diseñado para evaluar la seguridad y tolerabilidad de un inhibidor de la C1-esterasa en brotes desmielinizantes de NMO96.
ConclusionesLos datos revisados y comunicados en este artículo indican claramente que la NMO es una entidad completamente diferente a la EM. Tal es así que el dato más sobresaliente fue el descubrimiento de los anticuerpos anti-NMO, dado que contribuyó a la mejor caracterización de la NMO y su diferenciación con la EM97. Sin embargo, en muchas ocasiones la diferenciación se vuelve compleja en la práctica diaria. Durante las últimas 2 décadas se ha publicado variada información sobre la inmunopatogenia y tratamiento de esta entidad aunque mucho queda por investigar y descubrir. Actualmente diversas publicaciones avalan el tratamiento inmunosupresor preventivo de la NO o LETM en pacientes con IgG-NMO seropositivos para evitar un nuevo brote como se realiza en pacientes con síndrome clínico aislado de alto riesgo para EM (evitar EM clínicamente definida).
La complejidad diagnóstica en determinados pacientes es frecuente y aun con los criterios el diagnóstico se hace difícil. Por lo tanto, la historia clínica minuciosa y el examen neurológico son la base para el diagnóstico preciso en manos de neurólogos con experiencia en enfermedades desmielinizantes del SNC97.
