






El síndrome de Fisher-Bickerstaff forma parte del espectro clínico del síndrome anti-GQ1b. Se trata de un cuadro de polineuropatía inmunomediada, donde se evidencia un solapamiento entre el síndrome de Miller Fisher, caracterizado clínicamente por oftalmoplejía, ataxia y arreflexia, y la encefalitis de Bickerstaff, donde, a diferencia del anterior, existe déficit de vías largas con alteración del estado de consciencia, oftalmoplejía y ataxia. El diagnóstico se realiza sobre la en base de la presentación clínica, considerando que, dado el mecanismo patogénico común, el cuadro puede presentar características de ambas entidades.
Fisher- Bickerstaff syndrome is part of the clinical spectrum of the syndrome known as anti-GQ1b. It is an immune-mediated polyneuropathy, which showed overlap between Miller Fisher syndrome, characterized clinically by ophthalmoplegia, ataxia and areflexia, and Bickerstaff encephalitis, unlike the previous one where there is a shortage of long tracts with altered state of consciousness accompanying ophthalmoplegia and ataxia. The diagnosis will be made by the clinical presentation, considering that, given the common pathogenic mechanism may have characteristics of both entities.
El síndrome de Miller Fisher (SMF) ha sido considerado tradicionalmente como una variante clínica de la polirradiculoneuropatía inflamatoria idiopática aguda o síndrome de Guillain-Barré (SGB). Se caracteriza por la tríada de oftalmoplejía, ataxia y arreflexia; y fue descrito por Charles Miller Fisher en 19561 como diagnóstico diferencial de un evento isquémico agudo de tronco.
La encefalitis de tronco de Bickerstaff (EB) se caracteriza por alteración del estado de consciencia, ataxia, oftalmoplejía y signos de vías largas, tales como respuestas plantares extensoras o hemihipoestesia.
Estudios recientes identificaron que ambos síndromes se asocian a la presencia de anticuerpos anti-GQ1b y se ha propuesto que ambos cuadros clínicos conforman el espectro del síndrome de anticuerpos anti-GQ1b. Se ha propuesto denominar síndrome de Fisher-Bickerstaff a los cuadros que comparten características clínicas de ambas afecciones.
En cuanto al tratamiento de estas entidades, existe en la actualidad escasa evidencia sobre la indicación de la terapia con inmunoglobulina2, considerando el curso benigno de esta enfermedad3.
Se presenta el caso de una paciente con diagnóstico inicial de accidente cerebrovascular que presentó trastorno de la consciencia, ataxia, oftalmoplejía y arreflexia asociado a anticuerpos anti-GQ1b positivos. Asimismo, realizaremos una breve revisión de lo descrito en la literatura internacional.
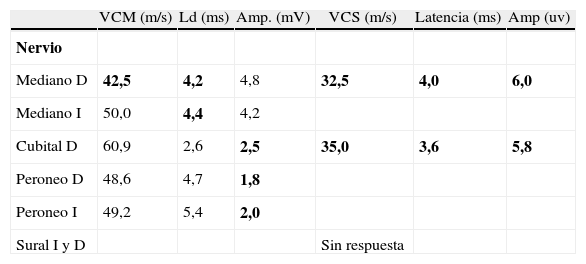
Caso clínicoMujer de 70 años, antecedentes de hipertensión arterial, cuadro gripal un mes previo al ingreso. Comienza en forma súbita con náuseas, vómitos, vértigo, disartria y deterioro del estado de consciencia. Es admitida en un hospital regional, diagnosticándose accidente vascular de tronco encefálico. Tres días después es dada de alta con remisión del cuadro de inicio, constatándose franca ataxia sensitiva para la marcha, leve ataxia cerebelosa en miembros, oftalmoparesia severa bilateral, diplopía, ptosis parcial derecha con pupilas isocóricas reactivas, parestesias distales en los 4 miembros, arreflexia global y sin debilidad muscular. Tomografía computarizada de encéfalo sin contraste: normal. Resonancia magnética (RM) y angio-RM muestra pequeñas imágenes de tipo microangiopático bihemisféricas, sin lesión en el tronco encefálico, con difusión negativa (fig. 1). El líquido cefalorraquídeo (LCR) evidencia disociación albuminocitológica (2 células por mm3 y proteínas de 70mg %; restantes valores, dentro de parámetros normales). El estudio de neuroconducción (tabla 1) muestra una disminución de la amplitud de los potenciales de acción muscular compuestos al explorar nervios peroneos de ambos lados y cubital derecho; latencias distales motoras levemente prolongadas de ambos nervios medianos, con disminución leve de la velocidad de conducción motora del nervio mediano derecho; latencias sensitivas de los nervios mediano derecho y cubital derecho prolongadas, con amplitudes y velocidades de conducción sensitivas disminuidas; sin respuesta al explorar ambos surales. Se realiza una determinación de anticuerpos antigangliósidos por el método de enzimoinmunoanálisis indirecto, con valores de referencia para los distintos gangliósidos: menor del 30%, negativo; de 31 a 50%, positivo débil; de 51 a 150%, positivo, y mayores de 150%, positivo fuerte. Presenta anti-GQ1b 111% (positivo) y anti-GD1a 50% (positivo débil), confirmando sospecha diagnóstica de síndrome de Fisher-Bickestraff. Inicia tratamiento con inmunoglobulina humana por vía intravenosa (IGIV), desarrollando a las 24 h paresia facial periférica izquierda y diplejía facial 48 h después, no progresando el cuadro con otros síntomas o signos de debilidad. Durante el curso del tratamiento con IGIV evoluciona sin parestesias y deambula sin ayuda, persistiendo leve inestabilidad, evidenciándose mejoría de la motilidad ocular en todas las direcciones, en especial en la mirada vertical.

Hallazgos de neuroconducción anormalidades
| VCM (m/s) | Ld (ms) | Amp. (mV) | VCS (m/s) | Latencia (ms) | Amp (uv) | |
| Nervio | ||||||
| Mediano D | 42,5 | 4,2 | 4,8 | 32,5 | 4,0 | 6,0 |
| Mediano I | 50,0 | 4,4 | 4,2 | |||
| Cubital D | 60,9 | 2,6 | 2,5 | 35,0 | 3,6 | 5,8 |
| Peroneo D | 48,6 | 4,7 | 1,8 | |||
| Peroneo I | 49,2 | 5,4 | 2,0 | |||
| Sural I y D | Sin respuesta |
Amp.: amplitud; D: derecho; I: izquierdo; Ld: latencia distal; VCM: velocidad de conducción motora; VN: valor normal; VCS: velocidad de conducción sensitiva. Los valores resaltados en “negrita” indican resultados anormales..
En 2001, Odaka et al. introdujeron el término «síndrome anti-GQ1b» para referirse a diferentes afecciones donde puede encontrarse el anticuerpo IgG anti-GQ1b4, indicando un mecanismo patogénico común.
El SMF, la oftalmoparesia aguda (OA), la EB y el SGB serían parte del espectro continuo de este sindrome5.
La patogénesis estaría explicada por mimetismo molecular, donde los anticuerpos generados contra diferentes microorganismos generan reacción cruzada con epítopos estructuralmente homólogos de los nervios o el tronco cerebral5,6. Esta teoría es apoyada por estudios epidemiológicos que evidenciaron antecedentes de infecciones bacterianas (Campylobacter jejuni7, Haemophilus influenzae8) y virales (citomegalovirus, virus de Epstein-Barr)9, tal como presentó nuestro paciente. Tanto en los pares iii, iv y vi (a nivel nodal y paranodal6), como en cerebelo y los ganglios de las raíces dorsales10, se detecta una mayor concentración en superficie celular del tetrasialogangliósido GQ1b, ligando de la IgG. Esto explicaría los hallazgos clínicos. El anticuerpo anti-GQ1b se liga también a la unión neuromuscular, causando una masiva liberación de acetilcolina desde las terminales nerviosas, produciendo bloqueo del terminal nervioso motor, alterando la transmisión6.
El SGB posee una incidencia mundial de 1-2 cada 100.000 habitantes11,12 y es la causa más frecuente de parálisis flácida de rápida instalación. El 25% inicia con parestesias ascendentes, presentándose dolor lumbar en el 50%11,12. Respecto del compromiso ventilatorio, el 30% evoluciona con requerimiento de asistencia ventilatoria11. Asimismo, es frecuente observar compromiso autonómico. El 50% de los pacientes desarrollan compromiso de pares craneanos, un 50% debilidad facial bilateral, un 50% debilidad orofaríngea y de un 10 a 20% cierto grado de compromiso ocular12,13. En ocasiones, se acompaña de edema de papila, posiblemente asociado a un déficit en la absorción del LCR consecuente a la hiperproteinorraquia13. Existen diferentes subtipos patológicos, siendo el más frecuente de desmielinización segmentaria multifocal. Considerado clásicamente como un cuadro monofásico, se describe hoy una recurrencia del 7% en un período promedio de 7 años14.
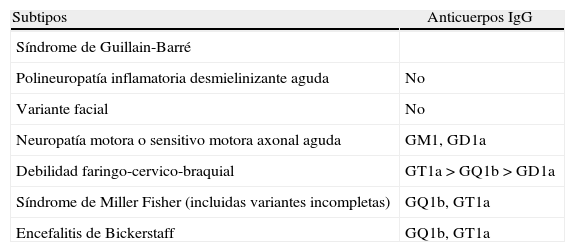
El SMF tiene una incidencia anual de alrededor de un paciente por millón15. Representa del 1 al 25% de todos los SGB y es más frecuente en hombres3. En un estudio reciente que incluyó a 19 pacientes, el síntoma de inicio fue diplopía en el 63%, mientras que la ataxia lo fue en un 5%. En todos hubo afectación del iii par, seguido por el vi (83%) y el iv (73%)16. En este síndrome el anticuerpo antigangliósido anti-GQ1b se encuentra en hasta el 90% de casos8 según las series, constituyendo parte de los criterios diagnósticos (tabla 2). La tríada característica no siempre se encuentra presente, apareciendo superposición con otras entidades del mismo síndrome. Se acompaña de midriasis en 50% de los casos17.
Principales anticuerpos según variante
| Subtipos | Anticuerpos IgG |
| Síndrome de Guillain-Barré | |
| Polineuropatía inflamatoria desmielinizante aguda | No |
| Variante facial | No |
| Neuropatía motora o sensitivo motora axonal aguda | GM1, GD1a |
| Debilidad faringo-cervico-braquial | GT1a>GQ1b>GD1a |
| Síndrome de Miller Fisher (incluidas variantes incompletas) | GQ1b, GT1a |
| Encefalitis de Bickerstaff | GQ1b, GT1a |
Fuente: Ropper12.
La EB es un cuadro caracterizado por oftalmoplejía externa progresiva, relativamente simétrica, acompañada por ataxia de al menos 4 semanas de evolución. Se asocia a signos de déficit de vías largas, junto con alteración del estado de consciencia18. Si bien las neuroimágenes no son patognomónicas, descartarían otros procesos, evidenciando en muchos casos imágenes hiperintensas en el tronco, el tálamo, el cerebelo y el cerebro18. Dos tercios de los pacientes con EB tienen anticuerpos IgG anti-GQ1b3,19.
La OA se define por oftalmoplejía externa, generalmente bilateral, sin ataxia. En algunos casos puede acompañarse también de midriasis, debido a presencia de antígeno GQ1b a nivel del ganglio ciliar o la placa terminal del músculo esfínter pupilar20.
En 2008, se sugirió el nuevo término «síndrome Fisher-Bickerstaff»18 para aquellos pacientes que, como el del caso descrito, evolucionaban en el curso de la enfermedad con oftalmoplejía, ataxia, reflejos osteotendinosos conservados y alteración del estado de consciencia21,22. Considerando que ambas entidades presentan un mecanismo fisiopatogénico común, basado en la presencia de autoanticuerpos anti-GQ1b, se ha propuesto que si la barrera hematoencefálica es defectuosa en ciertas regiones, como el área postrema, estos anticuerpos afectarían la formación reticular e inducirían la EB23.
El síndrome anti-GQ1b tiene un curso benigno, con recuperación espontánea24. Si bien en este caso se inició tratamiento con IGIV, existe escasa evidencia sobre la indicación de estas o plasmaféresis, aunque se ha reportado que ambas acortarían el tiempo de recuperación si son utilizadas precozmente2,24-28. El uso de estas terapias estaría justificado comprendiendo la entidad como un cuadro de autoinmunidad28.
Tal como se planteó con esta paciente y lo fue también en el primer caso reportado en la historia, uno de los principales diagnósticos diferenciales que se deben considerar es el accidente vascular de tronco encefálico (tabla 3). Teniendo en cuenta la nueva clasificación propuesta, podría entenderse el presente como un síndrome de Fisher-Bickerstaff, ya que reúne criterios diagnósticos de ambas entidades.
Diagnósticos diferenciales del síndrome de Fisher Bickerstaff
| Diagnóstico diferencial | Características |
| Botulismo | Causada por la neurotoxina botulínica tipo A, B, o E producida por Clostridium botulinum. Esta toxina actúa alterando la liberación presináptica, produciendo bloqueo neuromuscular. Puede evolucionar con compromiso de pares craneanos (diplopía, midriasis, parálisis facial). Puede haber alteración del habla y trastornos deglutorios. Con la progresión del cuadro: parálisis fláccida descendente hasta cuadriplejía, posible insuficiencia ventilatoria |
| Accidente vascular | Arteria cerebral posterior: infartos en territorio de distribución de ramas corticales: apraxia de movimientos oculares, déficit de campo visual. Infartos en territorios de ramas mesencefálicas: parálisis oculomotora homolateral con ataxia cerebelosa y temblor contralateral (síndrome de Claude)-parálisis oculomotora homolateral y hemiplejía contralateral (síndrome de Weber)-parálisis oculomotora homolateral, ataxia, temblor, coreoatetosis, hemiparesia contralateral (síndrome de Benedikt)-parálisis oculomotoraArteria basilar: deterioro del sensorio, déficit motor acompañado o no de déficit sensitivo en 4 MM, síndrome cerebeloso, afección de pares craneanos (parálisis de mirada lateral y/o vertical conjugada, diplopía, miosis). Síndromes protuberanciales: medial-parálisis de la mirada conjugada, diplopía, nistagmo, ataxiaInfartos cerebelosos: suelen asociarse a daño de tronco encefálico. Cuando el daño es cerebeloso «puro»: vértigo, náuseas, vómitos, cefalea, ataxia, paresia facial |
| Encefalopatía de Wernicke (déficit de tiamina) | Lesiones necróticas con pérdida de mielina a nivel de tronco, tálamo, hipotálamo, cerebelo, médula cervical. Lesiones de núcleos iii y vi |
| Miastenia gravis | Cuadro de curso crónico. Sintomatología motora que se caracteriza por fatigabilidad y fluctuación horaria: ptosis, diplopía, disartria, disfagia. Debilidad a predominio proximal |
Fuente: Yuki23.
Respecto a los hallazgos electrofisiológicos, a diferencia del SGB, donde la evidencia electrofisiológica de desmielinización segmentaria en el sistema nervioso periférico es frecuente, en el SMF los estudios de conducción nerviosa motora son normales en la mayoría de los casos, y si bien algunos autores reportan anomalías tales como la prolongación de las latencias distales, disminución de la velocidad de conducción nerviosa y disminución de la amplitud del potencial de acción muscular compuesto de grado leve (sin las clásicas características de las polineuropatías desmielinizantes adquiridas como dispersión temporal y bloqueos de conducción), los hallazgos electrofisiológicos más consistentes son la disminución de la amplitud de los potenciales de acción de nervios sensitivos fuera de proporción con prolongación de las latencias distales o disminución de la velocidad de conducción sensitiva, y la ausencia de reflejos H15,29–31.
Se postuló que los aferentes IA de los husos musculares están preferentemente afectados en el SMF y pueden ser responsables de los signos clínicos de ataxia y arreflexia, y la ausencia de reflejo H32,33. Respecto de las ondas F, los hallazgos son inconstantes y fueron reportadas series de pacientes con resultados dispares, hallando respuestas tardías F normales, prolongadas o ausentes15.
Se ha demostrado que los anticuerpos anti-GQ1b también causan daños estructurales en la unión neuromuscular, donde GQ1b se expresa en preparados de nervio-músculo del ratón34, y en la unión neuromuscular extraocular en el humano. Se postula que este daño a la unión neuromuscular en músculos extraoculares es responsable de la oftalmoplejía10. Usando electromiografía de fibra única, en pacientes con OA y anticuerpos anti-GQ1b elevados, se demostraron jitters anormales, que mejoraron con la recuperación clínica35, proporcionando la primera evidencia de defecto de la transmisión neuromuscular en pacientes con SMF.
La estimulación magnética transcraneal (EMT) es capaz de detectar disfunción corticoespinal subclínica. No sorprende que la EMT demuestre disfunción corticobulbar y corticoespinal en pacientes con la variante EB del síndrome de anticuerpos anti-GQ1b, donde también están presentes signos centrales36.
La presencia de anormalidades en los potenciales evocados (visuales, auditivos, somatosensitivos) ha proporcionado evidencia electrofisiológica de la combinación de lesiones centrales y periféricas en esta condición17,37.
Estos hallazgos subrayan el concepto de SMF/EB como un espectro continuo, con una combinación variable de anormalidades clínicas y subclínicas periféricas y centrales. SMF/EB parece ser un trastorno único del sistema nervioso, donde los signos clínicos y los hallazgos electrofisiológicos en consecuencia dependen de la expresión GQ1b en las estructuras afectadas: husos musculares, unión neuromuscular, nervios sensitivos, segmentos proximales de los nervios craneales15. Por todo lo expuesto, podemos concluir que el presente caso se trata de un síndrome de Fisher-Bickerstaff con presencia de anticuerpos anti-GQ1b, como se describió recientemente en la literatura. El mismo forma parte de un espectro continuo, compartiendo características clínicas de las distintas entidades que lo componen: SMF, SGB y EB. El descubrimiento de estos autoanticuerpos y su valor patogénico ha producido un avance en su conocimiento.
Es nuestra opinión que la búsqueda cuidadosa de síntomas o signos vinculados al sistema nervioso central durante el curso del síndrome de Fisher hará que este espectro sindrómico se pueda reconocer como una entidad nosológicamente definida.
Al Dr. Ricardo C. Reisin (Servicio de Neurología, Hospital Británico) por revisar y colaborar en la corrección del manuscrito; y a Orlando Gabriel Carballo y Silvia Graciela Ramos, bioquímicos del laboratorio de inmunología del Hospital Carlos G. Durand, por su ayuda en la comprensión de los materiales y métodos de los anticuerpos antigangliósidos.
