




La enfermedad de Fabry es el resultado de la deficiencia de alfa-galactosidasaA lisosomal, lo que resulta en depósito progresivo y patológico de glicoesfingolípidos en varias estirpes celulares. El dolor neuropático suele ser la primera manifestación en las variantes fenotípicas clásicas. Asociada a la insuficiencia renal y cardíaca, los accidentes cerebrovasculares pueden derivar en la muerte de los pacientes. Se describen los hallazgos de la evaluación clínica completa y de las neuroimágenes en 9 pacientes pertenecientes a una misma familia. Todos los pacientes presentaron acroparestesias y el 33% de los casos mostró lesiones isquémicas periventriculares silentes, así como en otro paciente se evidenciaron lesiones compatibles con un proceso inflamatorio. La enfermedad de Fabry debe ser incluida por los neurólogos dentro de las posibilidades diagnósticas en pacientes jóvenes con dolor neuropático (neuropatía de fibras finas) y en pacientes con stroke criptogénico debido a la posibilidad de un tratamiento específico que ha demostrado ser más eficaz cuanto más temprano sea indicado.
Fabry disease is a lysosomal storage disease caused by an inborn deficiency of alpha-galactosidaseA, which results in progressive accumulation of glycolipids in different cells and tissues. Neuropathic pain is usually the first symptom in classical variant. Chronic kidney disease and heart involvement associated with stroke are the main causes of death in Fabry patients. We present the complete assessment of clinical sign and symptoms and the neuroradiological evaluation in nine patients from a single family with Fabry disease. All cases showed neuropathic pain and 33% presented silent ischemic lesions in brain imaging, as well as also other case with multiple inflammatory findings. Fabry disease should be included by neurologists as part of screening in young patients with small fiber neuropathy and cryptogenic stroke due the chance of specific enzyme replacement therapy that showed better outcomes in early stages of the disease.
La enfermedad de Fabry (EF) es el resultado de la deficiencia de alfa-galactosidasaA (αGal-A), lo que resulta en depósito progresivo y patológico de glicoesfingolípidos1, predominantemente globotriaosilceramida (Gl3). Esta entidad, de herencia ligada al cromosomaX, tiene una incidencia de 1/40.000 nacidos vivos en su forma clásica de presentación, aunque a la fecha las variantes de inicio del adulto pueden ser tan frecuentes como 1/3.100 nacidos vivos2. El depósito de Gl3 se puede observar en células endoteliales, periteliales, neuronas, podocitos, cardiomiocitos, etc.1. Los primeros síntomas se expresan durante la niñez con dolor distal de tipo neuropático en los 4 miembros e hipohidrosis, asociado a lesiones cutáneas conocidas como angioqueratomas. Durante la adolescencia se agregan los depósitos de Gl3 en la córnea (córnea verticilada), nuevas manifestaciones disautonómicas, fatiga y disminución de la capacidad auditiva para, llegada la adultez, desarrollar insuficiencia renal y cardíaca, como también accidentes cerebrovasculares3.
Desde hace aproximadamente 13años la EF ha dado un giro significativo debido a la posibilidad de terapia de reemplazo enzimático (TRE), donde la enzima deficiente puede ser administrada en forma intravenosa4,5.
ObjetivoPresentar los hallazgos de la evaluación de la resonancia magnética cerebral y el compromiso multisistémico asociado en una familia con EF de la provincia de Córdoba, Argentina.
Pacientes y métodosSe evaluaron un total de 9 pacientes (6 hombres) con diagnóstico definitivo de EF por test bioquímicos y/o moleculares y pertenecientes a la misma familia. Solo 2 pacientes al momento de la evaluación eran menores a 18años: un niño de 16años y una niña de 14años. La presencia de compromiso cerebrovascular se evaluó con resonancia magnética cerebral (RMC) con resonador de 1,5T Signa Echo Speed (GE) System, con técnicas de adquisición estandarizadas para enfermedades cerebrovasculares (FLAIR, T1, T2 convencional, difusión y densidad protónica) y angiorresonancia de vasos intracraneanos sin contraste intravenoso. El examen clínico neurológico periférico evaluó la funcionalidad de las fibras finas (discriminación de la temperatura con tubos de agua fría y tibia y pinchazo) y fibras gruesas (reflejos osteotendinosos, parestesia y propiocepción). El examen multisistémico incluyó evaluación oftalmológica, nefrológica (examen de proteinuria y microalbuminuria de 24h e índice de filtrado glomerular medido con fórmula CKD-EPI) y cardiológica (examen ecocardiográfico y electrocardiográfico).
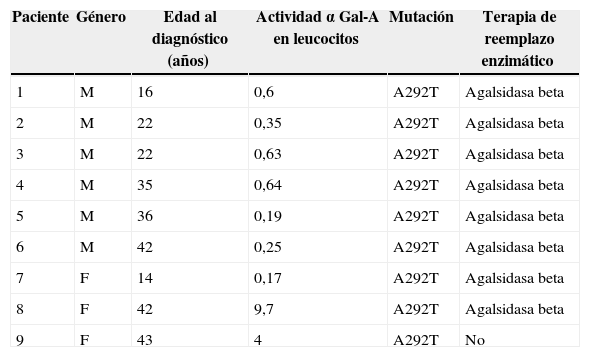
ResultadosUn total de 6 hombres (hemicigotos) y 3 mujeres (heterocigotas) fueron evaluados. Todos los pacientes presentaron niveles enzimáticos de αGal-A disminuidos (tabla 1). El estudio molecular reveló en todos los casos la mutación c.874G>A (p. Ala292Thr), ubicada en exón 6 del gen GLA que produce el cambio del aminoácido alanina por treonina. Esta mutación ha sido descrita previamente y se relaciona con el fenotipo clásico (severo) de la EF. Esto puede explicar la presencia de niveles enzimáticos muy por debajo del límite inferior en todas las heterocigotas, aun cuando se ha descrito la presencia de un 40% de falsos negativos con esta técnica diagnóstica6. Todos los pacientes hemicigotos y 2 de las 3 mujeres habían iniciado recientemente TRE con agalsidasa beta a 1mg/kg cada 14días.
Datos epidemiológicos. Actividad de α Gal-A en leucocitos (valor normal: 21-60 nmol/h/mg)
| Paciente | Género | Edad al diagnóstico (años) | Actividad α Gal-A en leucocitos | Mutación | Terapia de reemplazo enzimático |
|---|---|---|---|---|---|
| 1 | M | 16 | 0,6 | A292T | Agalsidasa beta |
| 2 | M | 22 | 0,35 | A292T | Agalsidasa beta |
| 3 | M | 22 | 0,63 | A292T | Agalsidasa beta |
| 4 | M | 35 | 0,64 | A292T | Agalsidasa beta |
| 5 | M | 36 | 0,19 | A292T | Agalsidasa beta |
| 6 | M | 42 | 0,25 | A292T | Agalsidasa beta |
| 7 | F | 14 | 0,17 | A292T | Agalsidasa beta |
| 8 | F | 42 | 9,7 | A292T | Agalsidasa beta |
| 9 | F | 43 | 4 | A292T | No |
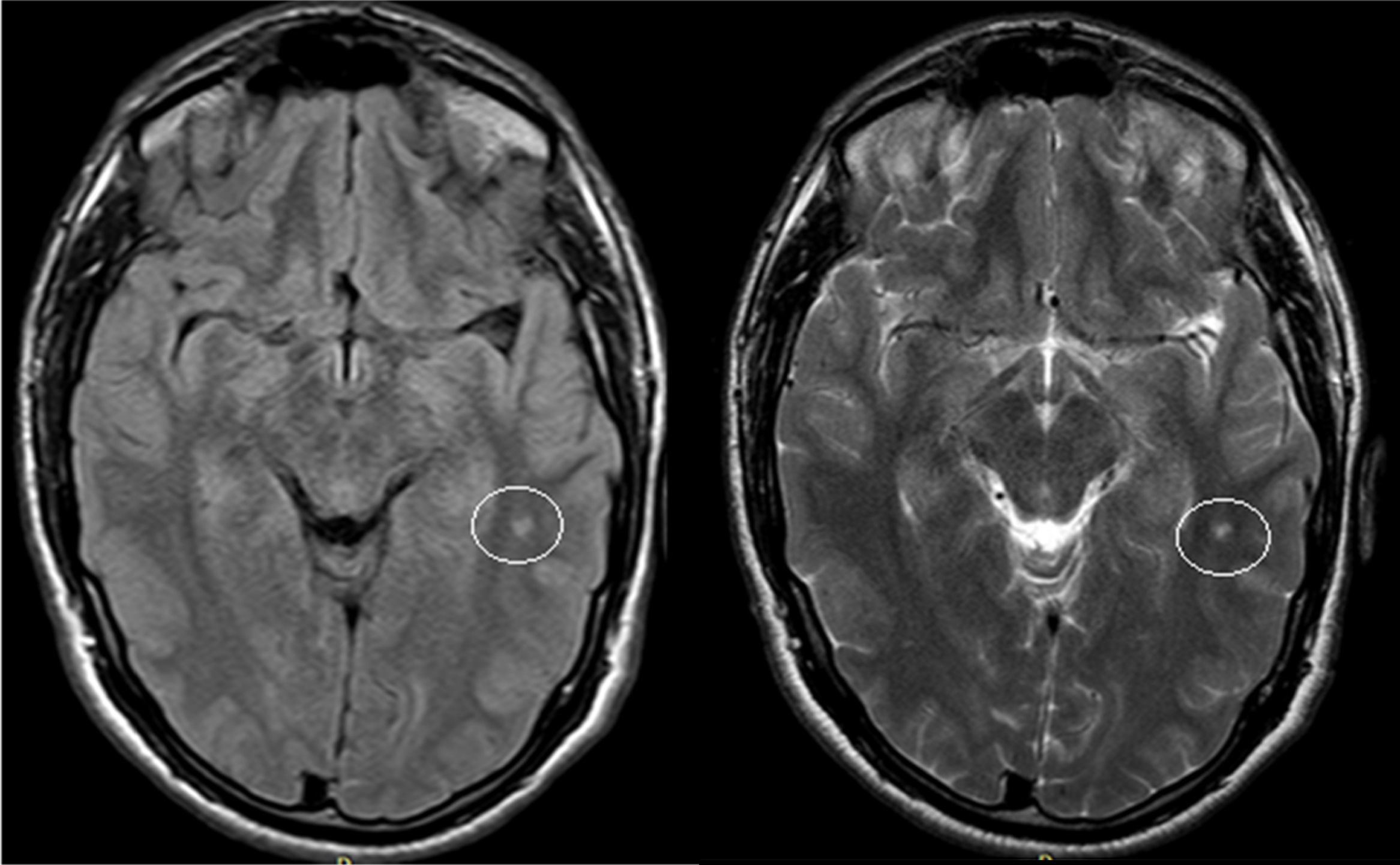
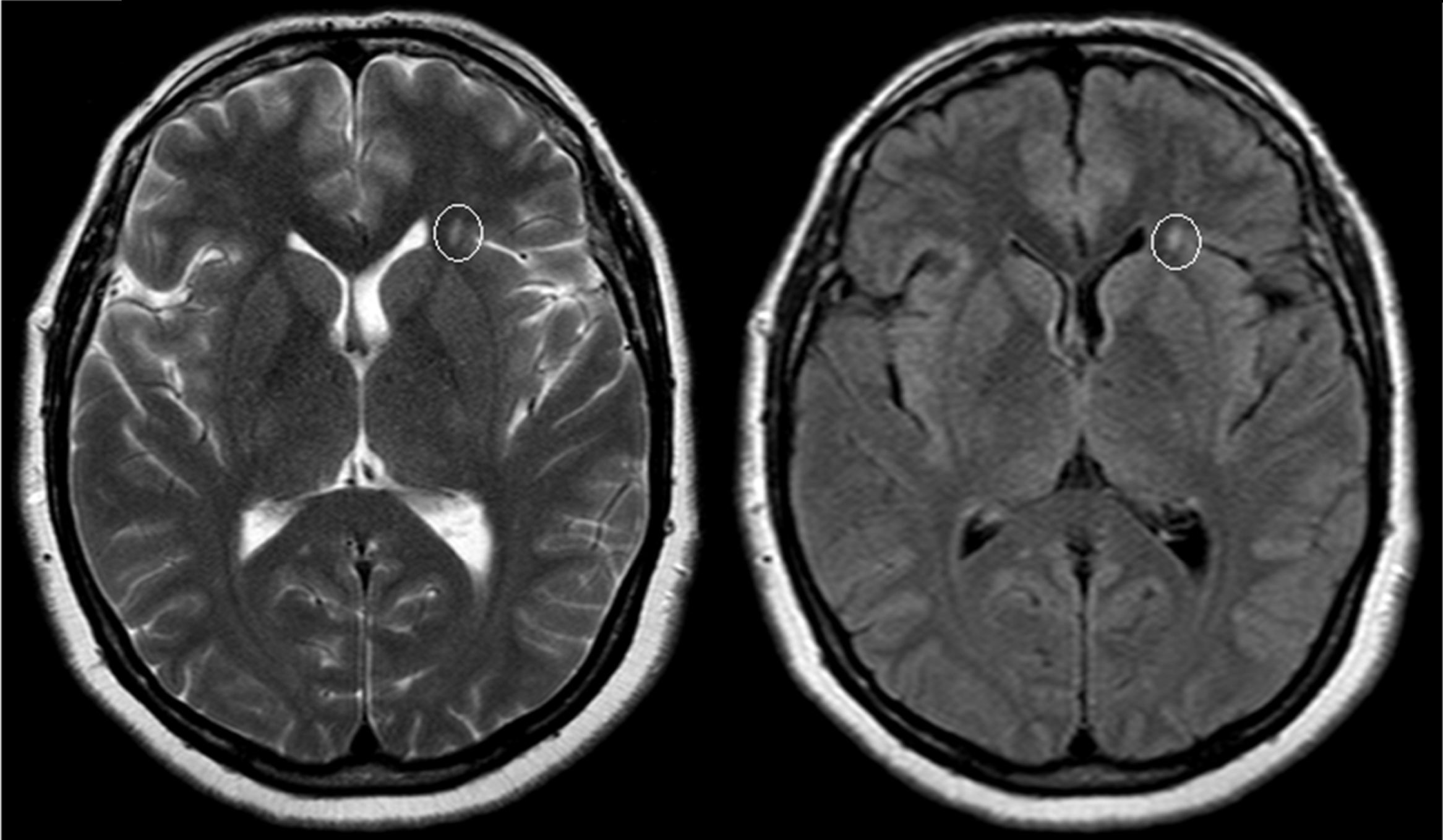
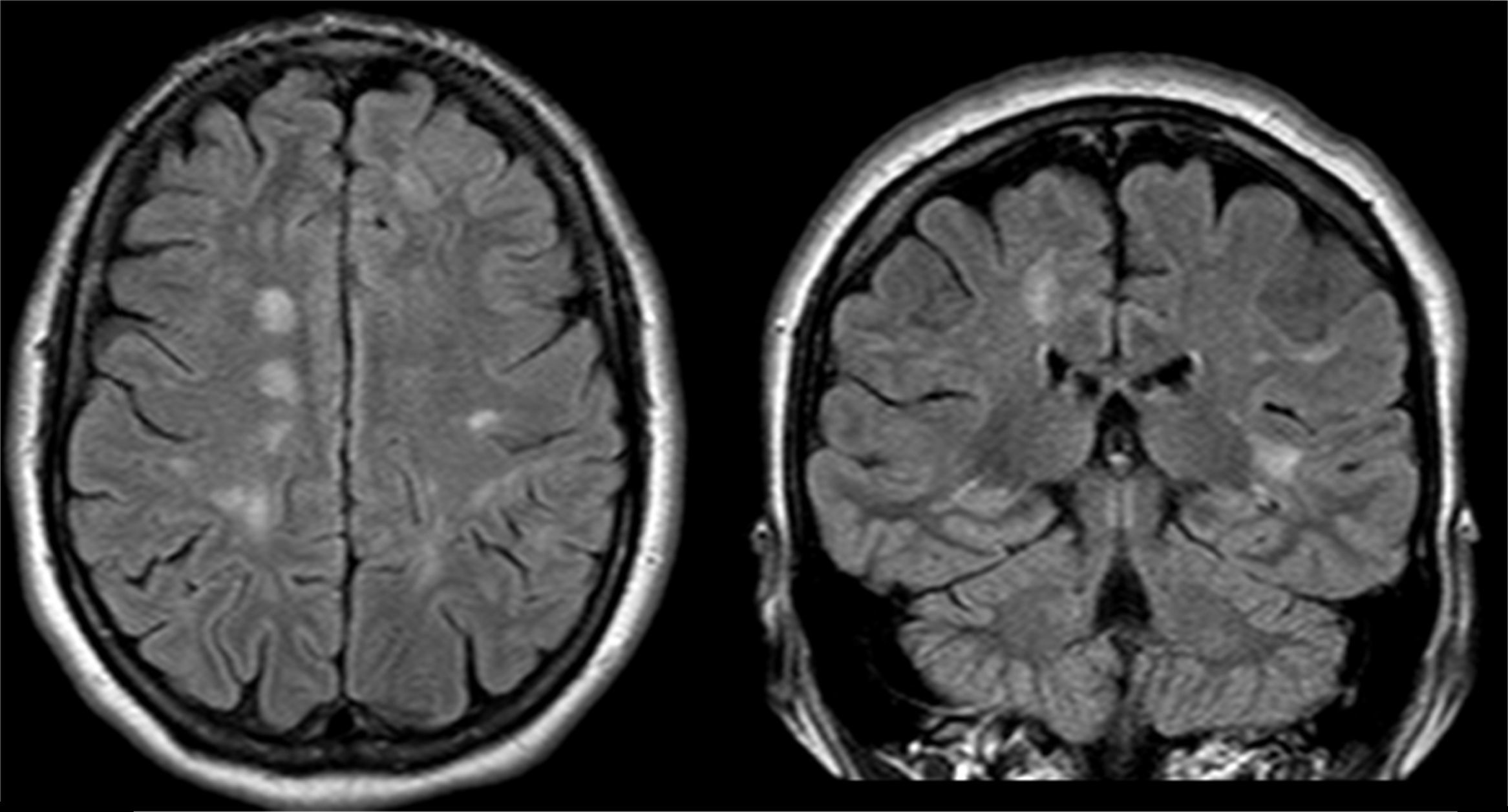
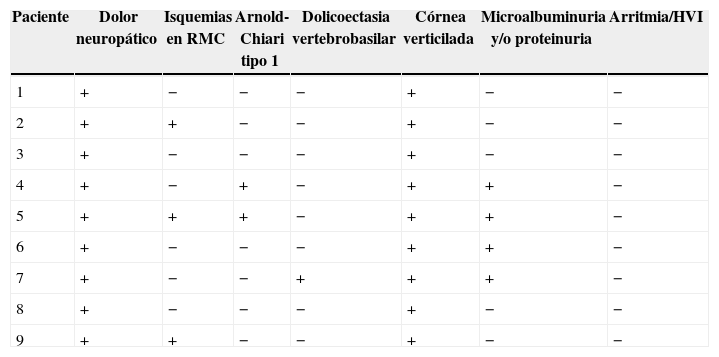
El examen clínico neurológico y la historia clínica no evidenciaron signos ni síntomas compatibles con la sospecha de daño cerebrovascular en nuestros pacientes. La evaluación de la RMC mostró lesiones isquémicas puntiformes periventriculares en 2 pacientes hemicigotos y una paciente heterocigota. La ausencia de compromiso clínico llevó a definir como lesiones «silentes» a estos hallazgos (tabla 2). Solo una paciente heterocigota presentó dolicoectasia vertebrobasilar y 2 pacientes hemicigotos malformación de Arnold-Chiari tipo1, siendo que otro mostró una lesión periventricular compatible con quiste neuroepitelial. Cabe destacar que una paciente sin signos clínicos de compromiso del sistema nervioso central y ausencia de las imágenes isquémicas previamente descritas en los otros pacientes, presentó múltiples imágenes supratentoriales bilaterales compatibles con lesiones inflamatorias diseminadas. El examen neurológico periférico no presentó anormalidades destacables, solo la presencia de disestesias típicas de la EF en todos los pacientes (comúnmente descritas en la literatura como acroparestesias) que aumentaban en intensidad durante la práctica de ejercicio, cambios de temperatura ambiental y fiebre.
Hallazgos de la evaluación neuroimagenológica y multisistémica
| Paciente | Dolor neuropático | Isquemias en RMC | Arnold-Chiari tipo 1 | Dolicoectasia vertebrobasilar | Córnea verticilada | Microalbuminuria y/o proteinuria | Arritmia/HVI |
|---|---|---|---|---|---|---|---|
| 1 | + | − | − | − | + | − | − |
| 2 | + | + | − | − | + | − | − |
| 3 | + | − | − | − | + | − | − |
| 4 | + | − | + | − | + | + | − |
| 5 | + | + | + | − | + | + | − |
| 6 | + | − | − | − | + | + | − |
| 7 | + | − | − | + | + | + | − |
| 8 | + | − | − | − | + | − | − |
| 9 | + | + | − | − | + | − | − |
HVI: hipertrofia del ventrículo izquierdo; RMC: resonancia magnética de cerebro.
El estudio cardiológico no mostró hipertrofia del ventrículo izquierdo en ninguno de los pacientes estudiados. La presencia de proteinuria fue evidente en 2 casos (un hemicigoto y una heterocigota) y valores compatibles con microalbuminuria (sin proteinuria) fueron hallados en 2 casos masculinos. El índice de filtrado glomerular se encontró dentro de los valores normales. El examen de lámpara de hendidura mostró córnea verticilada en todos los pacientes evaluados.
DiscusiónLa EF es una enfermedad progresiva que afecta a varios órganos y sistemas, y la TRE ha demostrado cambiar el curso natural de la enfermedad. Del análisis de las RMC se evidenciaron lesiones isquémicas en un tercio de la serie estudiada, lo que se encuentra dentro de los valores reportados con anterioridad (figs. 1 y 2). Si bien las lesiones en RMC suelen describirse luego de los 30años de edad7, algunos pacientes ya muestran lesiones isquémicas antes de los 20años. Uno de nuestros pacientes mostró una lesión lacunar a los 22años (fig. 2). Los mecanismos que se han reportado en relación a las lesiones cerebrovasculares son la presencia de estenosis luminal en pequeños vasos por depósito de Gl3 a nivel endotelial y de las células del músculo liso vascular (CMLV), factores promotores de hipertrofia celular muscular vascular, falta de autorregulación cerebrovascular y un posible factor protrombótico asociado a la EF3. Actualmente se ha descrito que el daño a nivel de las CMLV en grandes vasos cerebrales puede resultar en la presencia de dolicoectasia de predominio vertebrobasilar8. Este hallazgo puede ser aún más frecuente y precoz que la aparición de las típicas lesiones lacunares periventriculares. Solo una paciente mostró signos de dolicoectasia vertebrobasilar en esta serie. La malformación de Arnold-Chiari tipo1 ha sido descrita con mayor incidencia en la EF, no siendo clara la explicación de este hallazgo9. En nuestra serie hemos evidenciado 2 casos de malformación de Arnold-Chiari tipo1 sin presencia de la sintomatología clásicamente asociada a esta entidad. Si bien la fisiopatología de la EF no se ha relacionado con daño inflamatorio primario ni secundario a nivel renal, cardíaco ni cerebral, existen reportes donde se arribó a un diagnóstico final de coexistencia de EF y esclerosis múltiple u otras entidades relacionadas con neuroinflamación10,11. Previamente Lidove et al.12 reportaron algunos casos de meningoencefalopatía inflamatoria crónica, en base a los hallazgos en líquido cefalorraquídeo (LCR) y las características de las neuroimágenes. Estos hallazgos no han sido nuevamente descritos a la fecha. El patrón de imágenes hallado en una de nuestras pacientes puede ser descrito como compatible con lesiones inflamatorias (fig. 3). Debido a la ausencia total de síntomas, no se prosiguió con estudios de LCR, siendo planeado un control de RMC en 6meses.



Del análisis del compromiso multisistémico cabe destacar la alta prevalencia de córnea verticilada (100% de nuestra serie), situación que ha sido descrita previamente13. La ausencia de compromiso cardiológico posiblemente sea el hallazgo más discordante con la literatura en la actualidad.
El daño renal en la EF debe ser evaluado por medio de biopsia renal, ya que la ausencia de microalbuminuria y/o proteinuria no descarta el depósito de Gl3 a nivel podocitario, endotelial, mesangial y tubular. Las biopsias renales efectuadas en pacientes pediátricos sin proteinuria ni caída del índice de filtrado glomerular muestran compromiso podocitario y borramiento concomitante de los pedicelos, situación que indefectiblemente resultará en proteinuria y progresión del daño renal14. Estos hallazgos han resultado en cambios de los paradigmas en la fisiopatología de la EF, siendo que la presencia de borramiento de pedicelos en la biopsia renal es criterio suficiente para iniciar la TRE, aun en ausencia de proteinuria. Nuestra serie mostró microalbuminuria y/o proteinuria en el 45% de los casos.
Actualmente 8 de los 9 pacientes se encuentran bajo TRE con agalsidasa beta en dosis de 1mg/kg cada 14días. Esta terapia ha sido aprobada en Europa, Japón, Latinoamérica y Estados Unidos. Los primeros estudios que llevaron a la aprobación de la agalsidasa beta para el tratamiento de la EF en el año 2001 se realizaron con pacientes con escasa afección renal y fueron excluidos quienes presentaban una creatinina sérica mayor de 2,2mg/dl, necesidad de diálisis o trasplante renal. Esta población mostró que luego de 5meses de tratamiento con dosis de 1mg/kg se logró una remoción completa del Gl3 del endotelio capilar renal, cuantificado por medio de biopsia renal previa y posterior al tratamiento4. Estos resultados fueron reproducidos y mantenidos luego de 54meses de tratamiento15.
La EF debe ser incluida por los neurólogos dentro de las posibilidades diagnósticas en pacientes jóvenes con dolor neuropático (neuropatía de fibras finas) y en pacientes con stroke criptogénico debido a la posibilidad de un tratamiento específico que ha demostrado ser más eficaz cuanto más temprano sea indicado.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que los procedimientos seguidos se conformaron a las normas éticas del comité de experimentación humana responsable y de acuerdo con la Asociación Médica Mundial y la Declaración de Helsinki.
Confidencialidad de los datosLos autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores han obtenido el consentimiento informado de los pacientes y/o sujetos referidos en el artículo. Este documento obra en poder del autor de correspondencia.
Conflicto de interesesTodos los autores han aprobado la versión final del manuscrito y no tienen conflictos de intereses que declarar.
